Analysis of mutational signatures with musicatk in the R console
Aaron Chevalier, Joshua Campbell
Compiled December 16, 2024
Source:vignettes/articles/tutorial_tcga_cnsl.Rmd
tutorial_tcga_cnsl.RmdIntroduction
A variety of exogenous exposures or endogenous biological processes
can contribute to the overall mutational load observed in human tumors.
Many different mutational patterns, or “mutational signatures”, have
been identified across different tumor types. These signatures can
provide a record of environmental exposure and can give clues about the
etiology of carcinogenesis. The Mutational Signature Comprehensive
Analysis Toolkit (musicatk) contains a complete end-to-end workflow for
characterization of mutational signatures in a cohort of samples.
musicatk has utilities for extracting variants from a variety of file
formats, multiple methods for discovery of novel signatures or
prediction of pre-existing signatures, and many types of downstream
visualizations for exploratory analysis. This package has the ability to
parse and combine multiple motif classes in the mutational signature
discovery or prediction processes. Mutation motifs include single base
substitutions (SBS), double base substitutions (DBS), insertions (INS)
and deletions (DEL). The package can be loaded using the
library command:
Importing mutational data
In order to discover or predict mutational signatures, we must first set up our musica object by 1) extracting variants from files or objects such as VCFs and MAFs, 2) selecting the appropriate reference genome 3) creating a musica object, 4) adding sample-level annotations, and 5) building a count tables for our variants of interest. Alternatively, a musica object can be created directly from a count table.
Import variants from files
Variants can be extracted from various formats using the following functions:
- The
extract_variants_from_vcf_file()function will extract variants from a VCF file. The file will be imported using the readVcf function from the VariantAnnotation package and then the variant information will be extracted from this object. - The
extract_variants_from_vcf()function extracts variants from aCollapsedVCForExpandedVCFobject from the VariantAnnotation package. - The
extract_variants_from_maf_file()function will extract variants from a file in Mutation Annotation Format (MAF) used by TCGA. - The
extract_variants_from_maf()function will extract variants from a MAF object created by the maftools package. - The
extract_variants_from_matrix()function will get the information from a matrix or data.frame like object that has columns for the chromosome, start position, end position, reference allele, mutation allele, and sample name. - The
extract_variants()function will extract variants from a list of objects. These objects can be any combination of VCF files, VariantAnnotation objects, MAF files, MAF objects, and data.frame objects.
Below are some examples of extracting variants from MAF and VCF files:
# Extract variants from a MAF File
lusc_maf <- system.file("extdata", "public_TCGA.LUSC.maf", package = "musicatk")
lusc.variants <- extract_variants_from_maf_file(maf_file = lusc_maf)
# Extract variants from an individual VCF file
luad_vcf <- system.file("extdata", "public_LUAD_TCGA-97-7938.vcf",
package = "musicatk"
)
luad.variants <- extract_variants_from_vcf_file(vcf_file = luad_vcf)
# Extract variants from multiple files and/or objects
melanoma_vcfs <- list.files(system.file("extdata", package = "musicatk"),
pattern = glob2rx("*SKCM*vcf"), full.names = TRUE
)
variants <- extract_variants(c(lusc_maf, luad_vcf, melanoma_vcfs))Import TCGA datasets
For this tutorial, we will analyze mutational data from lung and skin
tumors from TCGA. This data will be retrieved using the the
GDCquery function from TCGAbiolinks
package.
library(TCGAbiolinks)
tcga_datasets <- c("TCGA-LUAD", "TCGA-LUSC", "TCGA-SKCM")
types <- gsub("TCGA-", "", tcga_datasets)
variants <- NULL
annot <- NULL
for (i in seq_along(tcga_datasets)) {
# Download variants
query <- GDCquery(
project = tcga_datasets[i],
data.category = "Simple Nucleotide Variation",
data.type = "Masked Somatic Mutation",
workflow.type = "Aliquot Ensemble Somatic Variant Merging and Masking",
experimental.strategy = "WXS",
data.format = "maf"
)
GDCdownload(query)
data <- GDCprepare(query)
# Extract from maf
temp <- extract_variants_from_matrix(data)
variants <- rbind(variants, temp)
annot <- rbind(annot, cbind(
rep(types[i], length(unique(temp$sample))),
unique(as.character(temp$sample))
))
}
colnames(annot) <- c("Tumor_Type", "ID")
rownames(annot) <- annot[, "ID"]Note that with previous versions of the GDC database, you may need to
set worflow.type to another string such as
workflow.type = MuTect2 Variant Aggregation and Masking.
Creating a musica object
A genome build must first be selected before a musica object can be
created for mutational signature analysis. musicatk uses BSgenome
objects to access genome sequence information that flanks each mutation
which is used bases for generating mutation count tables. BSgenome
objects store full genome sequences for different organisms. A full list
of supported organisms can be obtained by running
available.genomes() after loading the BSgenome library.
Custom genomes can be forged as well (see BSgenome
documentation). musicatk provides a utility function called
select_genome() to allow users to quickly select human
genome build versions “hg19” and “hg38” or mouse genome builds “mm9” and
“mm10”. The reference sequences for these genomes are in UCSC format
(e.g. chr1).
g <- select_genome("hg38")The last preprocessing step is to create an object with the variants
and the genome using the create_musica_from_variants
function. This function will perform checks to ensure that the
chromosome names and reference alleles in the input variant object match
those in supplied BSgenome object. These checks can be turned off by
setting check_ref_chromosomes = FALSE and
check_ref_bases = FALSE, respectively. This function also
looks for adjacent single base substitutions (SBSs) and will convert
them to double base substitutions (DBSs). To disable this automatic
conversion, set convert_dbs = FALSE.
musica <- create_musica_from_variants(x = variants, genome = g)## Checking that chromosomes in the 'variant' object match chromosomes in the 'genome' object.## Checking that the reference bases in the 'variant' object match the reference bases in the 'genome' object.## Standardizing INS/DEL style## Removing 22 compound insertions## Removing 167 compound deletions## Converting 44 insertions## Converting 246 deletions## Converting adjacent SBS into DBS## 15176 SBS converted to DBSImporting sample annotations
Sample-level annotations, such as tumor type, treatment, or outcome
can be used in downstream analyses. Sample annotations that are stored
in a vector or data.frame can be directly
added to the musica object using the
samp_annot function:
id <- as.character(sample_names(musica))
samp_annot(musica, "Tumor_Type") <- annot[id, "Tumor_Type"]Note: Be sure that the annotation vector or data.frame being supplied is in the same order as the samples in the
musicaobject. Thesample_namesfunction can be used to get the order of the samples in the musica object. Note that the annotations can also be added later on.
Creating mutation count tables
Create standard tables
Motifs are the building blocks of mutational signatures. Motifs themselves are a mutation combined with other genomic information. For instance, SBS96 motifs are constructed from an SBS mutation and one upstream and one downstream base sandwiched together. We build tables by counting these motifs for each sample.
build_standard_table(musica, g = g, table_name = "SBS96")## Building count table from SBS with SBS96 schemaHere is a list of mutation tables that can be created by setting the
table_name parameter in the
build_standard_table function:
- SBS96 - Motifs are the six possible single base pair mutation types times the four possibilities each for upstream and downstream context bases (464 = 96 motifs)
- SBS192_Trans - Motifs are an extension of SBS96 multiplied by the
transcriptional strand (translated/untranslated), can be specified with
"Transcript_Strand". - SBS192_Rep - Motifs are an extension of SBS96 multiplied by the
replication strand (leading/lagging), can be specified with
"Replication_Strand". - DBS - Motifs are the 78 possible double-base-pair substitutions
- INDEL - Motifs are 83 categories intended to capture different categories of indels based on base-pair change, repeats, or microhomology, insertion or deletion, and length.
Combine tables
Different count tables can be combined into one using the
combine_count_tables function. For example, the SBS96 and
the DBS tables could be combined and mutational signature discovery
could be performed across both mutations modalities. Tables with
information about the same types of variants (e.g. two related SBS
tables) should generally not be combined and used together.
# Build Double Base Substitution table
build_standard_table(musica, g = g, table_name = "DBS78")## Building count table from DBS with DBS78 schema
# Combine with SBS table
combine_count_tables(musica, to_comb = c("SBS96", "DBS78"), name = "SBS_DBS",
description =
"An example combined table, combining SBS96 and DBS")
# View all tables
names(tables(musica))## [1] "SBS96" "DBS78" "SBS_DBS"Creating a musica object directly from a count table
If a count table is already available, a musica object can be created directly without need for a variant file and building tables.
luad_count_table_path <- system.file("extdata", "luad_tcga_count_table.csv",
package = "musicatk"
)
luad_count_table <- as.matrix(read.csv(luad_count_table_path))
musica_from_counts <- create_musica_from_counts(luad_count_table, "SBS96")Filtering samples
Samples with low numbers of mutations should usually be excluded from
discover and prediction procedures. The
subset_musica_by_counts function can be used to exclude
samples with low numbers of mutations in a particular table:
musica_filter <- subset_musica_by_counts(musica, table_name = "SBS96",
num_counts = 10)The subset_musica_by_annotation function can also be
used to subset the musica object to samples that match a particular
annotation. For example, if we only wanted to analyze lung cancer, we
could filter to samples that have “LUAD” or “LUSC”:
musica_luad <- subset_musica_by_annotation(musica, annot_col = "Tumor_Type",
annot_names = c("LUAD", "LUSC"))Discovery of signatures and exposures
Mutational signature discovery is the process of deconvoluting a
matrix containing the count of each mutation type in each sample into
two matrices: 1) a Signature matrix containing the
probability of each mutation motif in signature and 2) an
Exposure matrix containing the estimated counts of each
signature in each sample. Discovery and prediction results are saved in
the result_list slot of a musica object. The
discover_signatures function can be used to identify
signatures in a dataset de novo.
Determining an appropriate k value
The k value is the number of signatures that are predicted from a
discovery method. To help determine an appropriate k value, the
compare_k_vals function can be used to compare to the
stability and error associated with various k values. Generally, 100
replicates is suggested, as well as a larger span of k values to test.
Here, fewer k values and replicates are used for simplicity.
k_comparison <- compare_k_vals(musica, "SBS96",
reps = 100, min_k = 2, max_k = 6,
algorithm = "lda"
)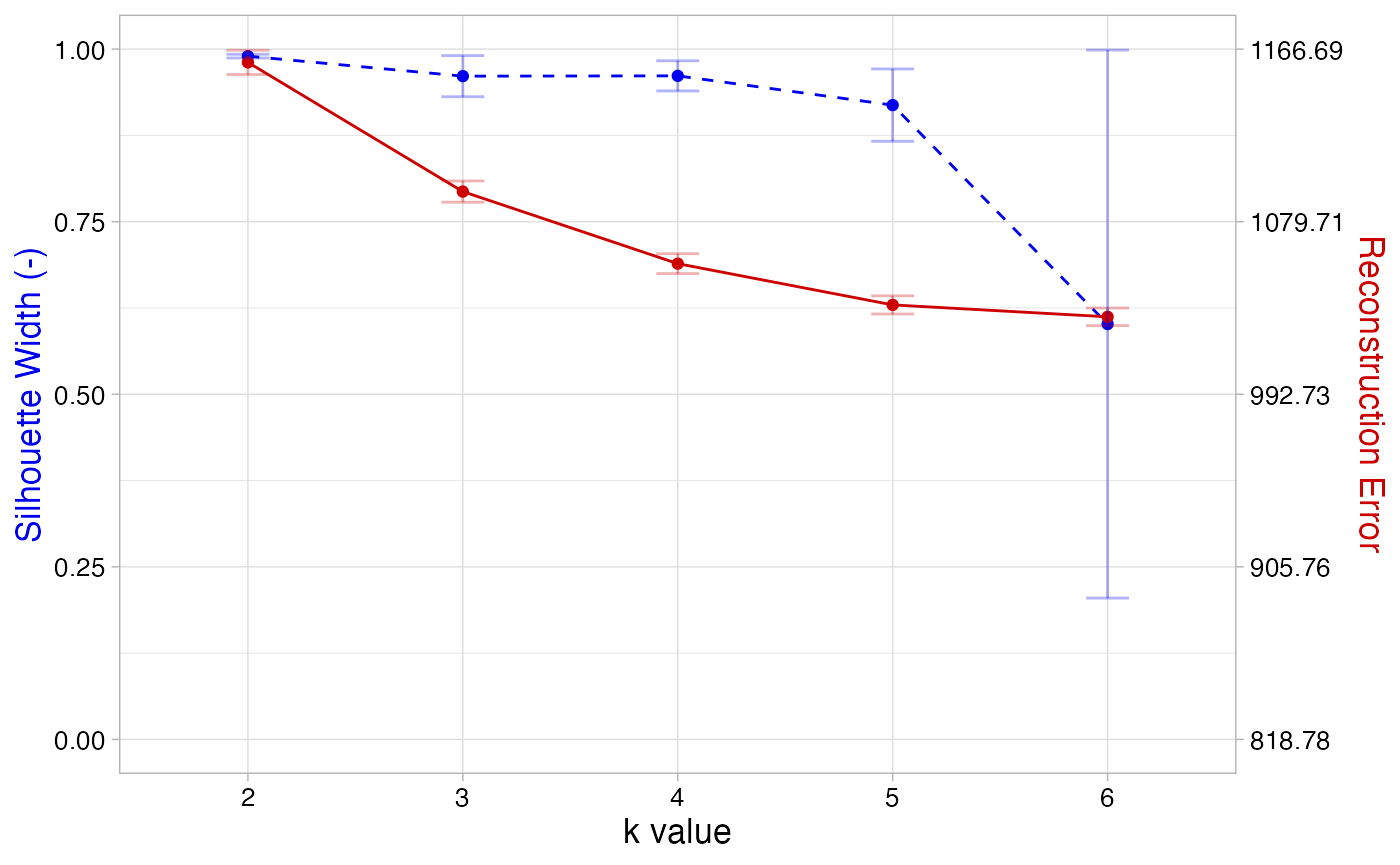
From the resulting plot, we see that k = 4 yields a relatively high
silhouette width and a relatively low reconstruction error. The error
bars for both metrics are also quite narrow for k = 4. Therefore four
signatures should be selected, so we will set k equal to 4 in the
discover_signatures function:
discover_signatures(musica_filter,
modality = "SBS96", num_signatures = 4,
algorithm = "lda", model_id = "ex_result"
)Supported signature discovery algorithms include:
- Non-negative matrix factorization (nmf)
- Latent Dirichlet Allocation (lda)
Both have built-in seed capabilities for reproducible
results, nstarts for multiple independent chains from which
the best final result will be chosen. NMF also allows for parallel
processing via par_cores. To get the signatures or
exposures from the result object, the following functions can be
used:
# Extract the exposure matrix
expos <- exposures(musica_filter, "result", "SBS96", "ex_result")
expos[1:3, 1:3]## TCGA-99-8033-01A-11D-2238-08 TCGA-99-8032-01A-11D-2238-08
## Signature1 106.52551 285.88771
## Signature2 44.75862 53.23718
## Signature3 7.65405 10.74107
## TCGA-99-8028-01A-11D-2238-08
## Signature1 370.2086953
## Signature2 21.8723410
## Signature3 0.8796583
# Extract the signature matrix
sigs <- signatures(musica_filter, "result", "SBS96", "ex_result")
sigs[1:3, 1:3]## Signature1 Signature2 Signature3
## C>A_ACA 0.03277323 0.004306005 5.521593e-04
## C>A_ACC 0.02966502 0.005984863 3.379481e-04
## C>A_ACG 0.02146643 0.002121666 1.519013e-05Visualization of results
Plot signatures
The plot_signatures function can be used to display
barplots that show the probability of each mutation type in each
signature:
plot_signatures(musica_filter, "ex_result")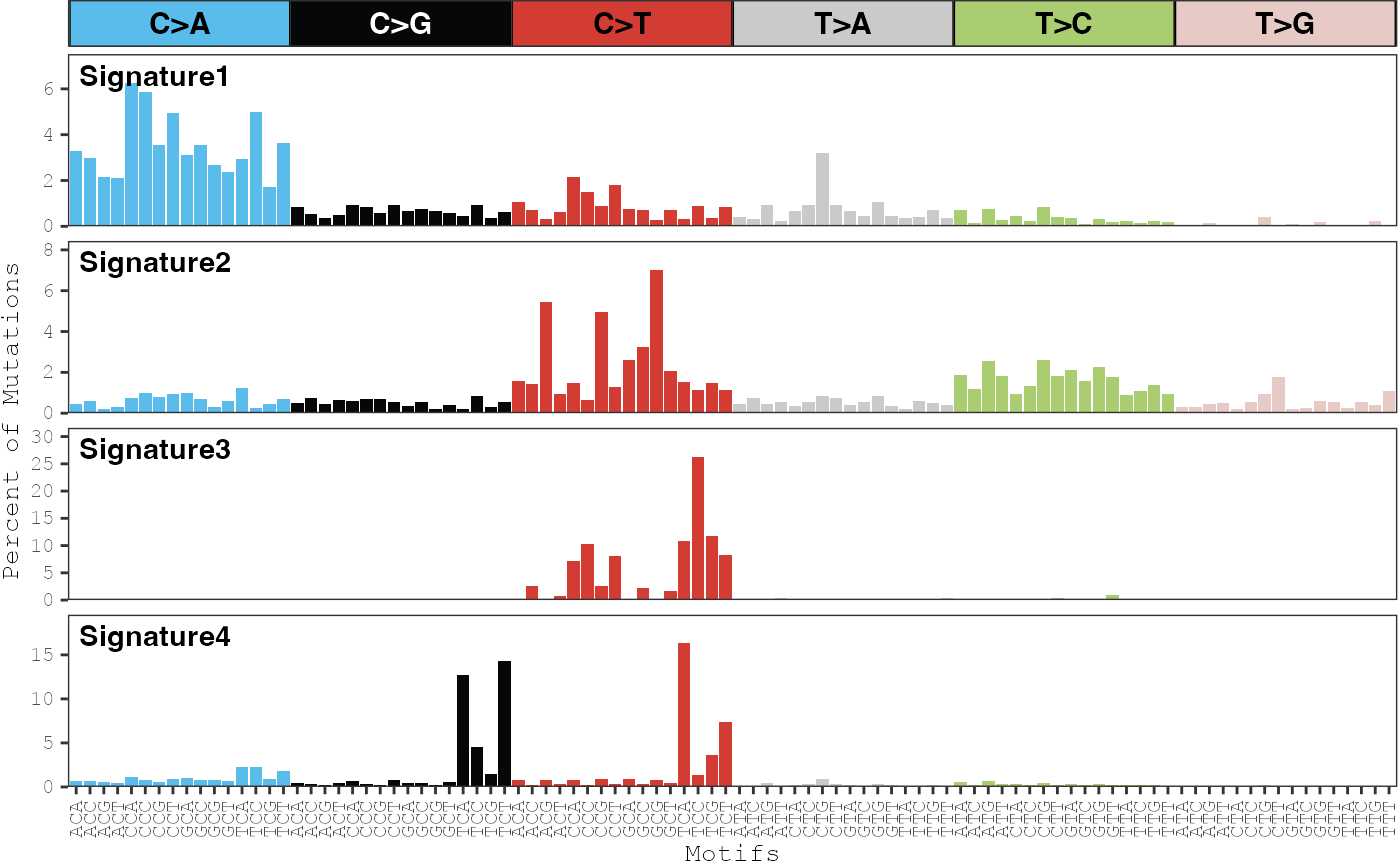
By default, the scales on the y-axis are forced to be the same across
all signatures. This behavior can be turned off by setting
same_scale = FALSE:
plot_signatures(musica_filter, "ex_result", same_scale = FALSE)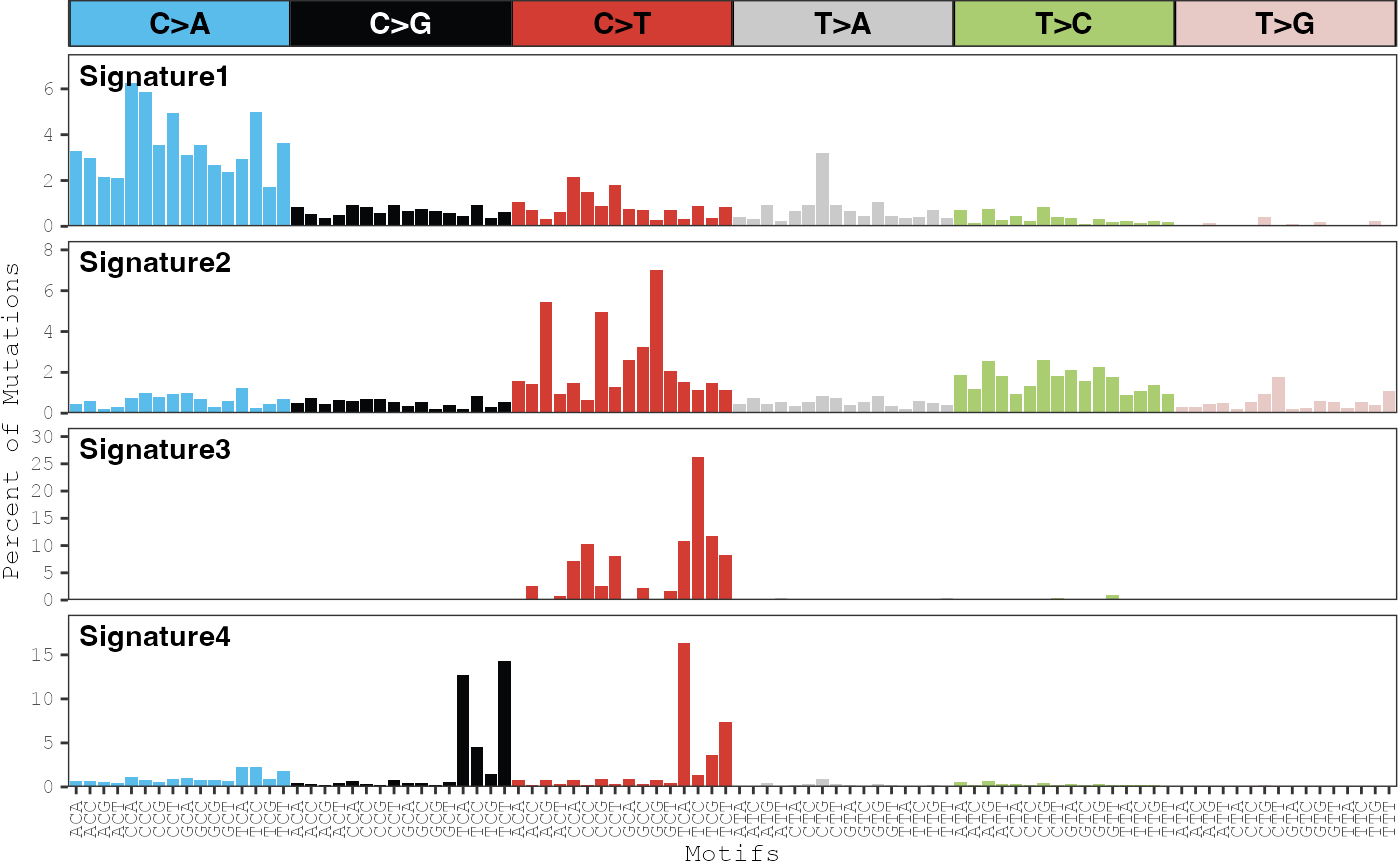
Comparing to external signatures
A common analysis is to compare the signatures estimated in a dataset
to those generated in other datasets or to those in the COSMIC
database. We have a set of functions that can be used to easily
perform pairwise correlations between signatures. The
compare_results functions compares the signatures between
two models in the same or different musica objects. The
compare_cosmic_v2 will correlate the signatures between a
model and the SBS signatures in COSMIC V2. For example:
compare_cosmic_v2(musica_filter, "ex_result", threshold = 0.8)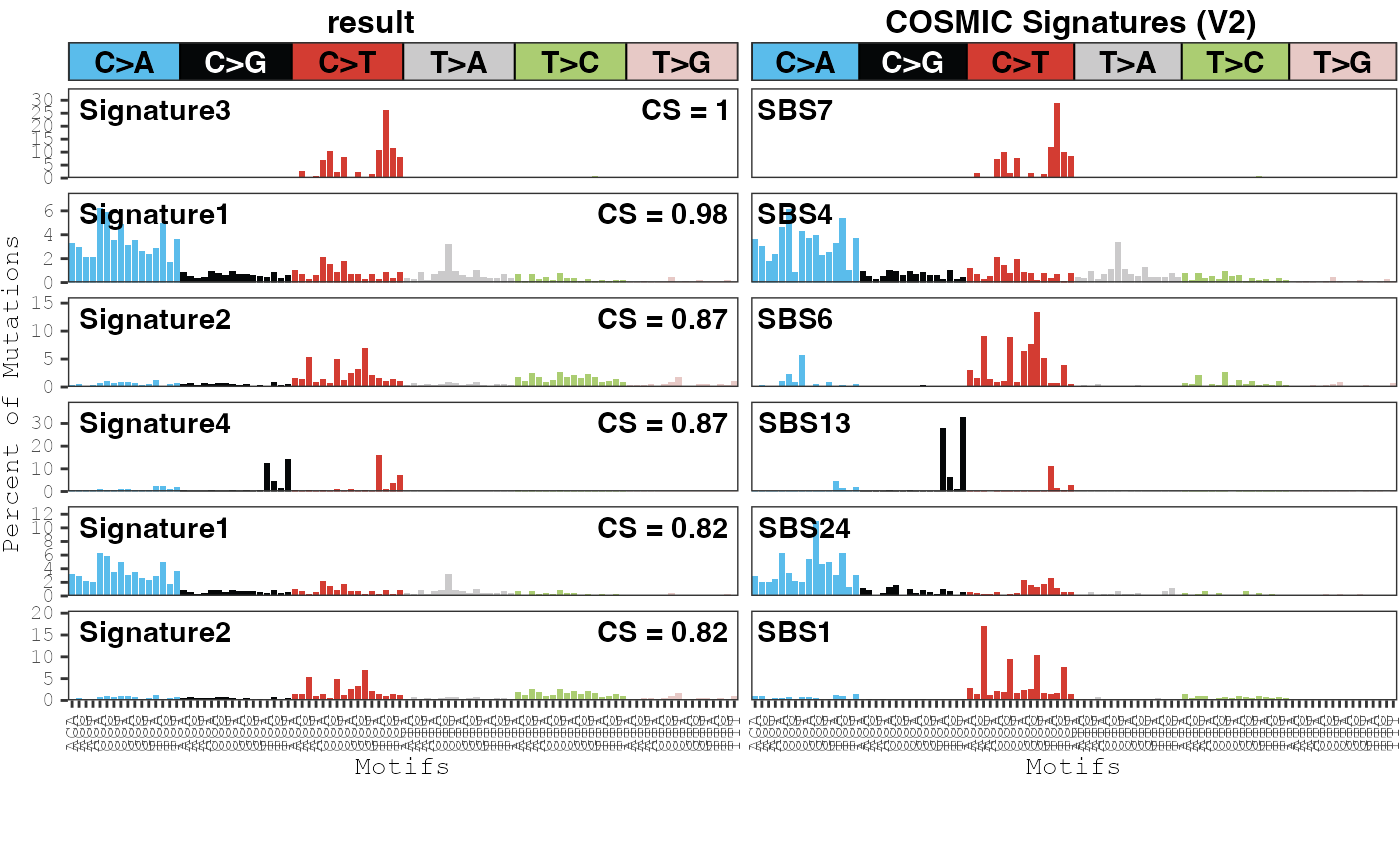
## cosine x_sig_index y_sig_index x_sig_name y_sig_name
## 5 0.9964997 3 7 Signature3 SBS7
## 1 0.9753047 1 4 Signature1 SBS4
## 4 0.8698311 2 6 Signature2 SBS6
## 6 0.8679439 4 13 Signature4 SBS13
## 2 0.8207367 1 24 Signature1 SBS24
## 3 0.8165057 2 1 Signature2 SBS1In this example, our Signatures 1 and 3 were most highly correlated
to COSMIC Signature 4 and 7, respectively, so this may indicate that
samples in our dataset were exposed to UV radiation or cigarette smoke.
Only pairs of signatures who have a correlation above the
threshold parameter will be returned. If no pairs of
signatures are found, then you may want to consider lowering the
threshold. Signatures can also be correlated to those in the COSMIC V3
database using the compare_cosmic_v3 function.
Based on the COSMIC comparison results and our prior knowledge, these signatures can be re-named and the new name can displayed in the plots:
name_signatures(musica_filter, "ex_result",
c("SBS4 - Smoking", "SBS15 - MMR", "SBS7 - UV",
"SBS2/13 - APOBEC"))
plot_signatures(musica_filter, "ex_result")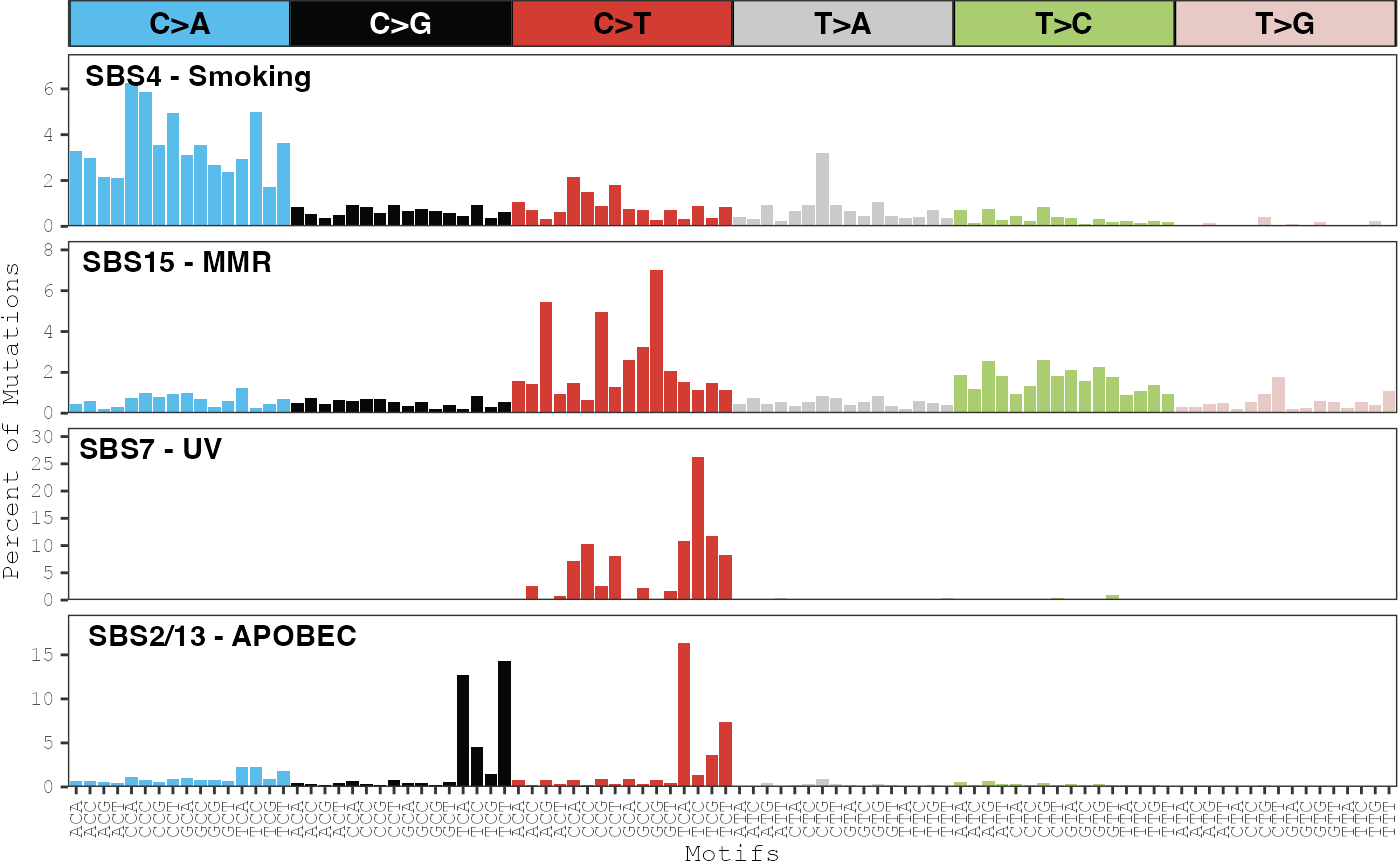
Plot exposures
Barplots showing the exposures in each sample can be plotted with the
plot_exposures function:
plot_exposures(musica_filter, "ex_result", plot_type = "bar")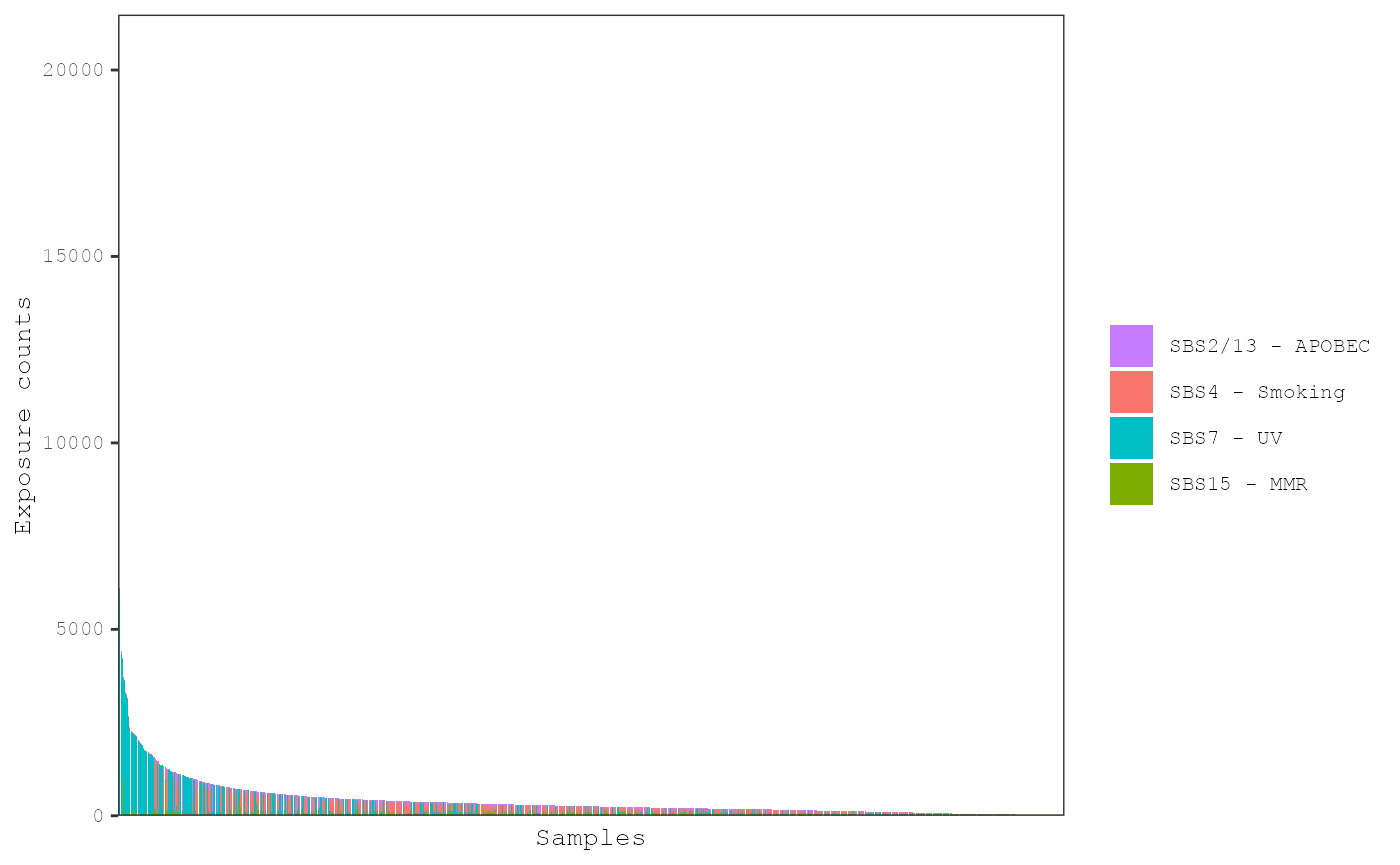
By default, samples are ordered from those with the highest number of
mutations on the left to those with the lowest on the right. Sometimes,
too many samples are present and the bars are too small to clearly
examine the patterns of exposures. The num_samples
parameter can be used to display the top samples with the highest number
of mutations on the left:
plot_exposures(musica_filter, "ex_result", plot_type = "bar", num_samples = 50)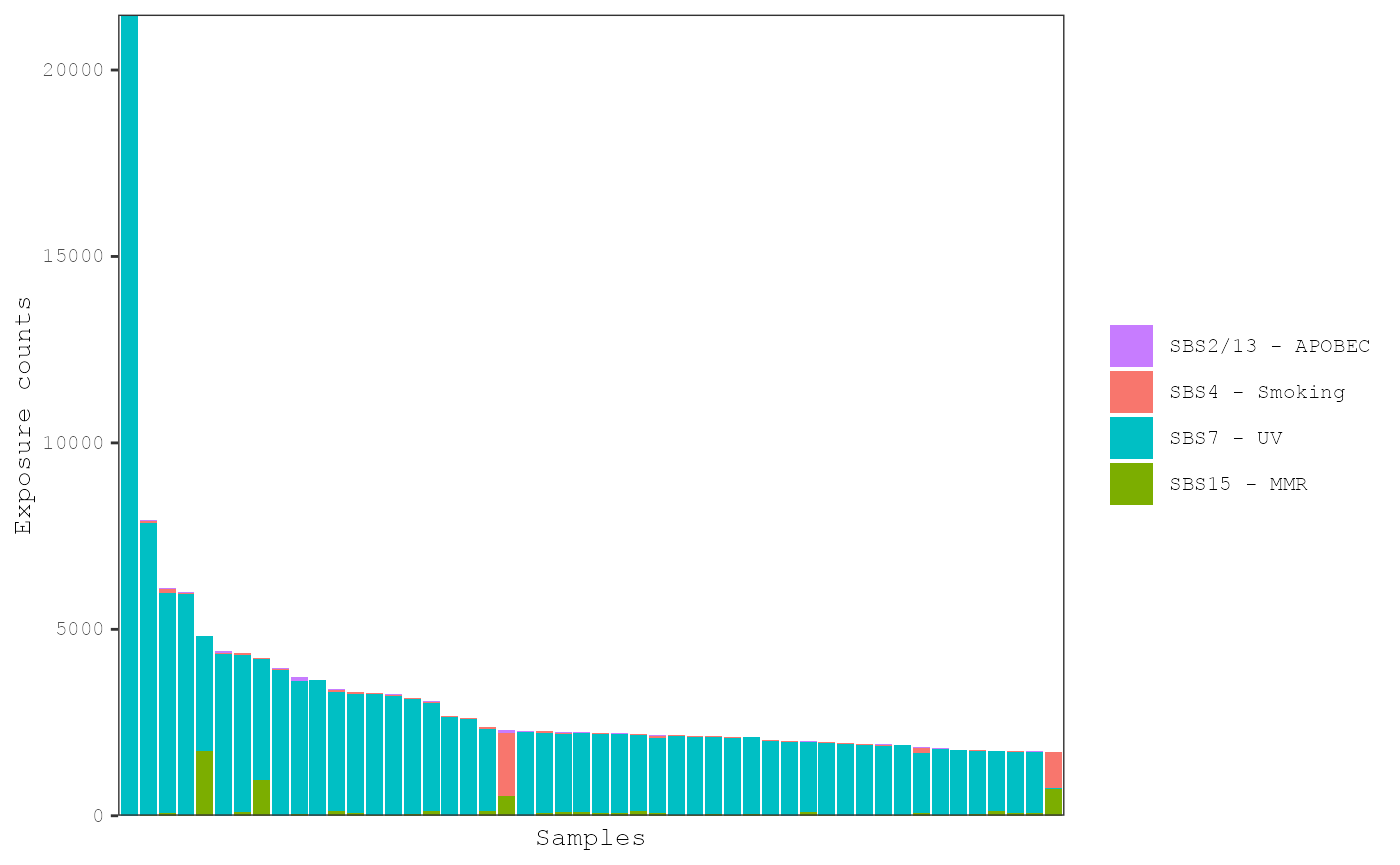 Samples can be ordered by the level of individual exposures. The can be
used in combination with the
Samples can be ordered by the level of individual exposures. The can be
used in combination with the num_samples parameter to
examine the mutational patterns in the samples with the highest levels
of a particular exposure. For example, samples can be ordered by the
number of estimated mutations from the MMR signature:
plot_exposures(musica_filter, "ex_result", plot_type = "bar", num_samples = 50,
sort_samples = "SBS15 - MMR")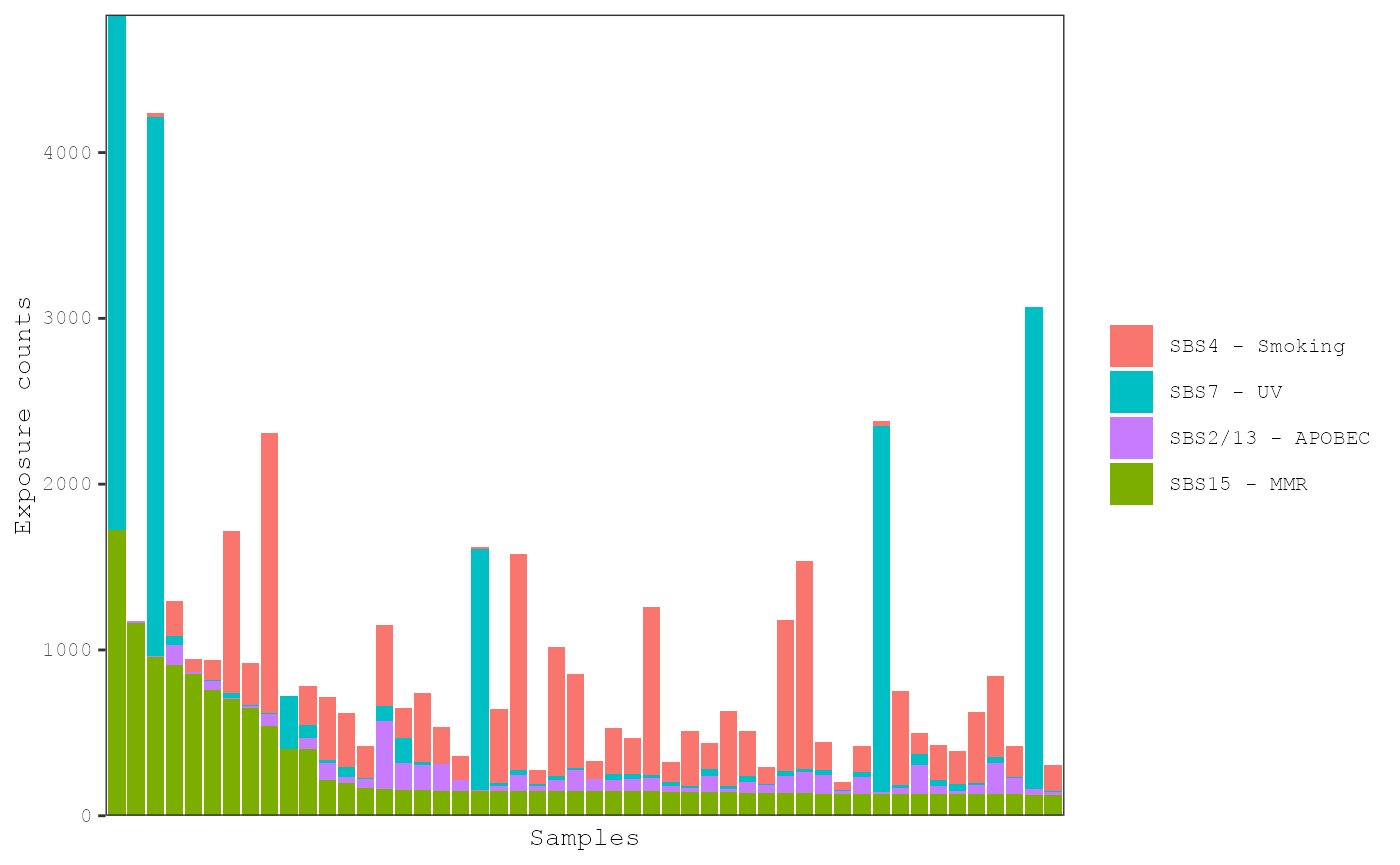
The proportion of each exposure in each tumor can be shown by setting
proportional = TRUE:
plot_exposures(musica_filter, "ex_result", plot_type = "bar", num_samples = 50,
proportional = TRUE)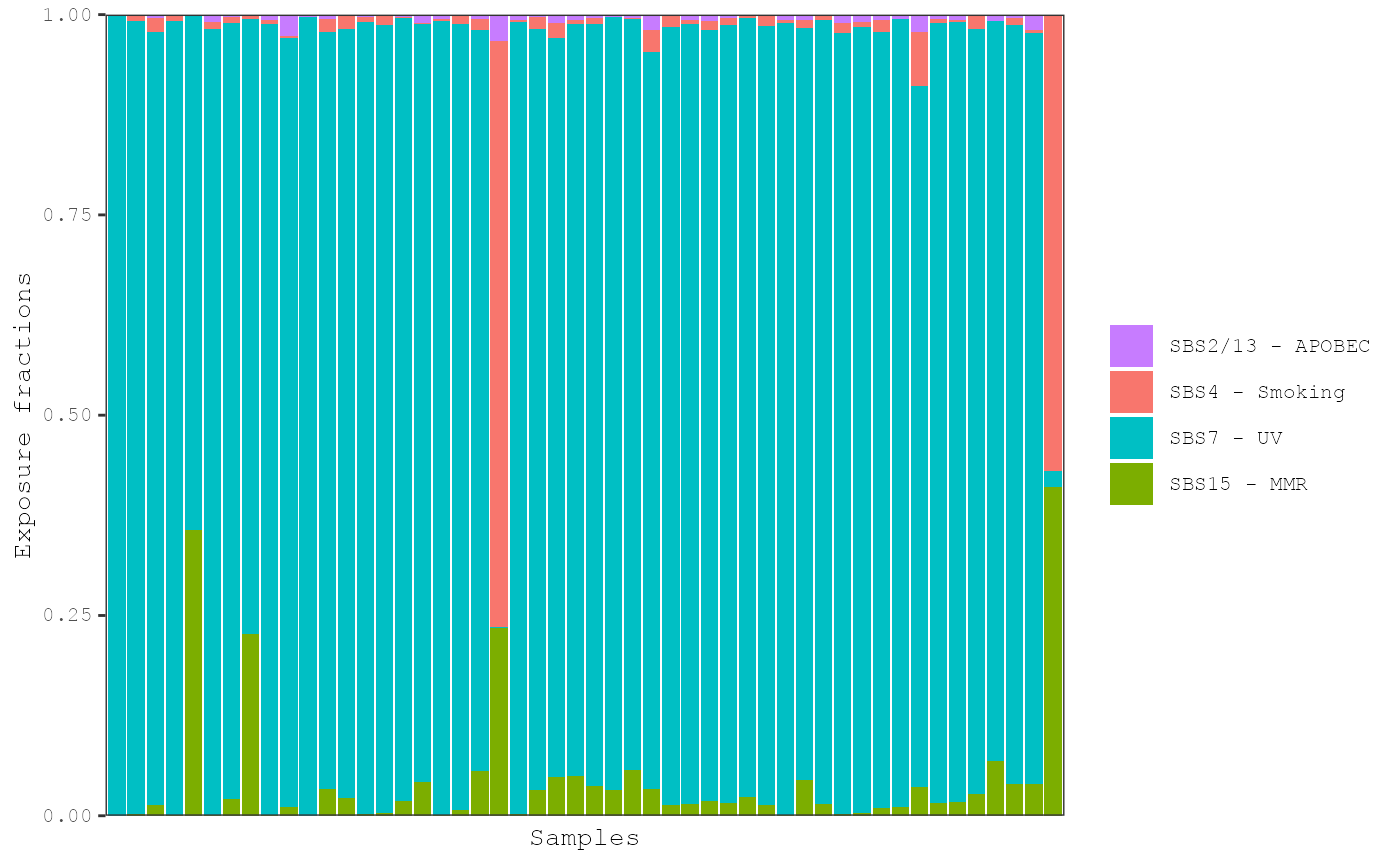
The plot_exposures function can group exposures by
either a sample annotation or by a signature by setting the
group_by parameter. To group by an annotation, the
groupBy parameter must be set to "annotation"
and the name of the annotation must be supplied via the
annotation parameter. For example, the exposures from the
previous result can be grouped by the Tumor_Type
annotation:
plot_exposures(musica_filter, "ex_result", plot_type = "bar",
group_by = "annotation", annotation = "Tumor_Type")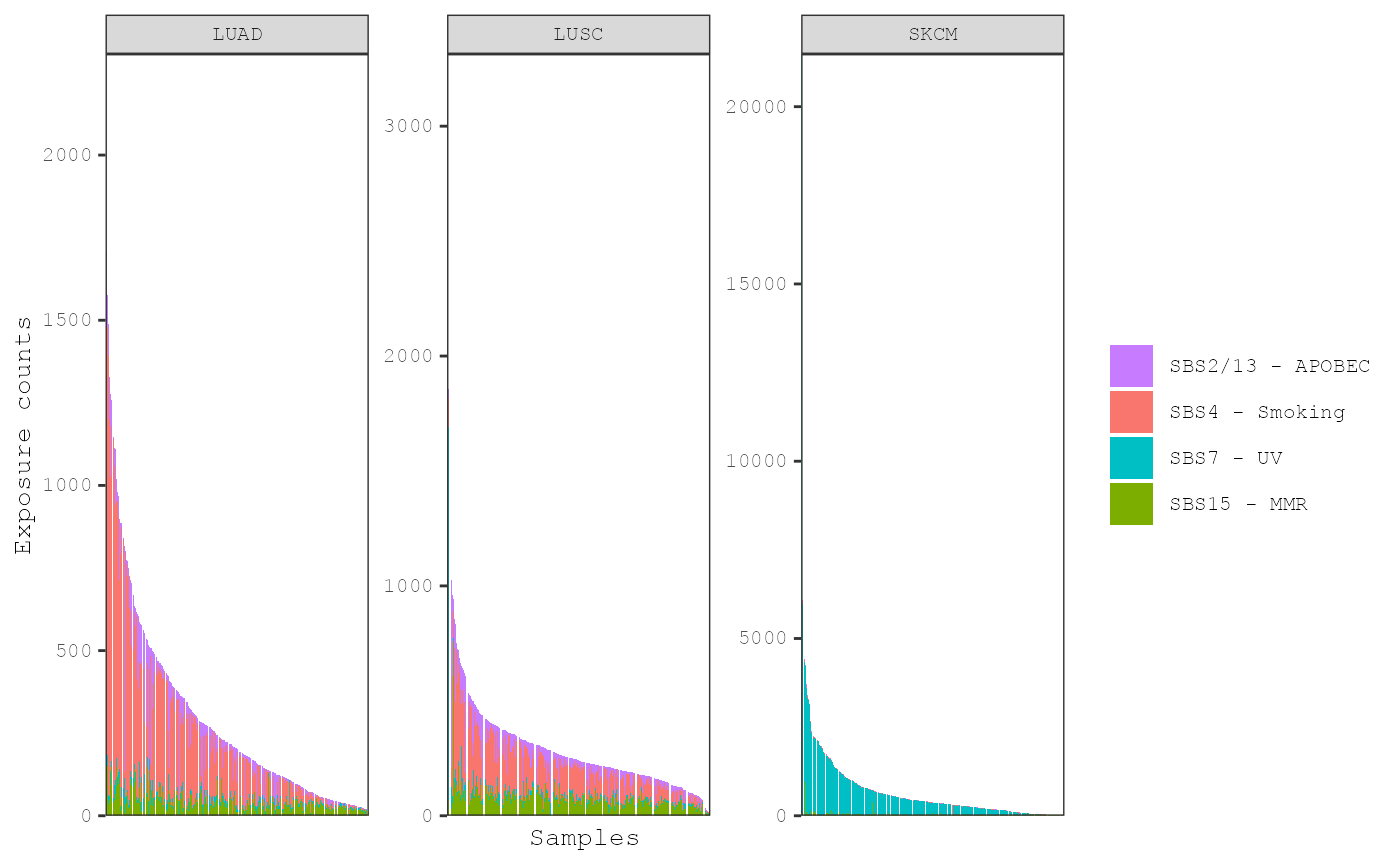
In this plot, it is clear that the smoking signature is more active
in the lung cancers while the UV signature is more active in the skin
cancers. The distribution of exposures with respect to annotation can be
viewed using boxplots by setting plot_type = "box" and
group_by = "annotation":
plot_exposures(musica_filter, "ex_result", plot_type = "box",
group_by = "annotation", annotation = "Tumor_Type")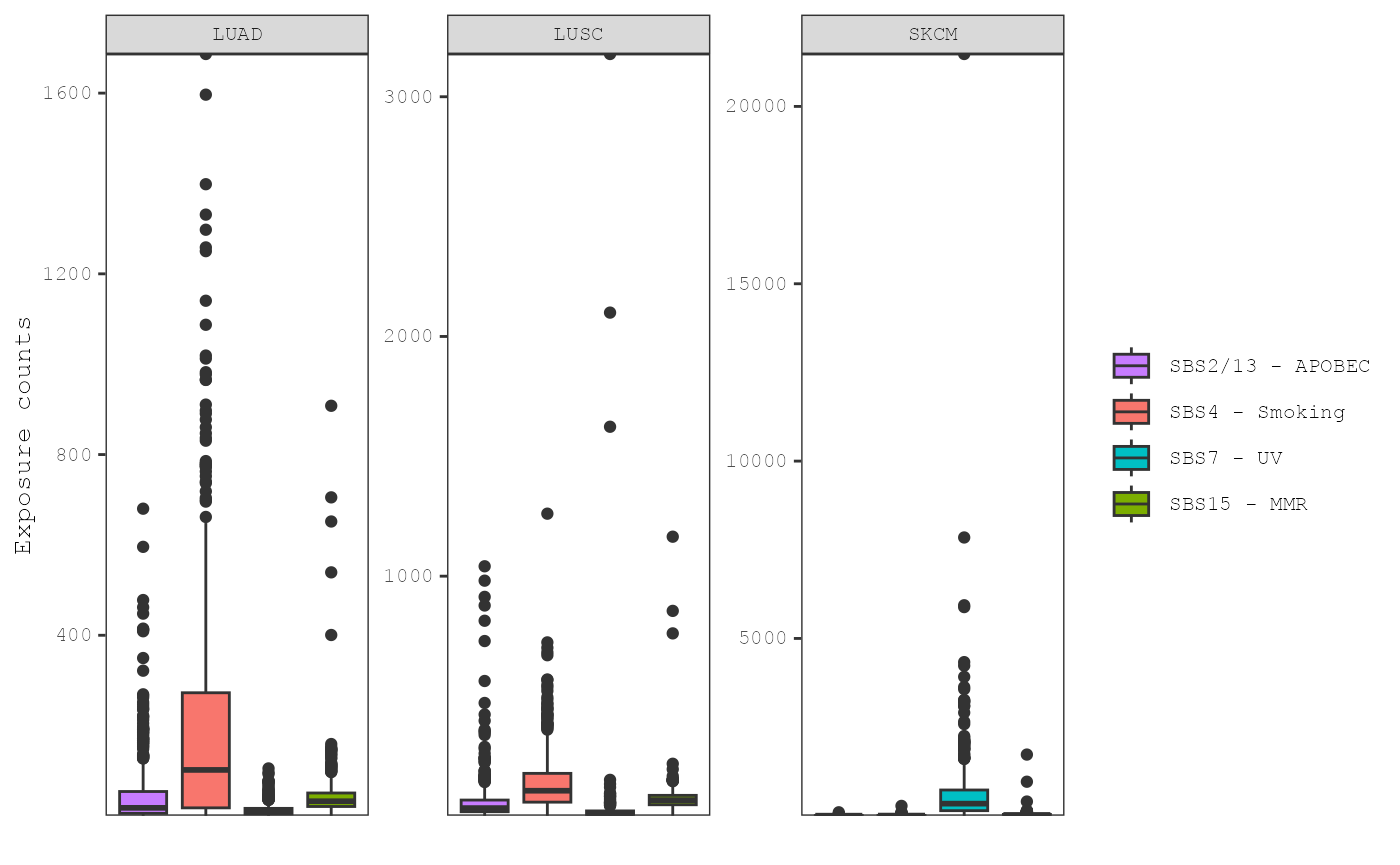
Note that boxplots can be converted to violin plots by setting
plot_type = "violin". To compare the exposures levels
across groups of samples within a signature, we can set
group_by = "signature" and
color_by = "annotation":
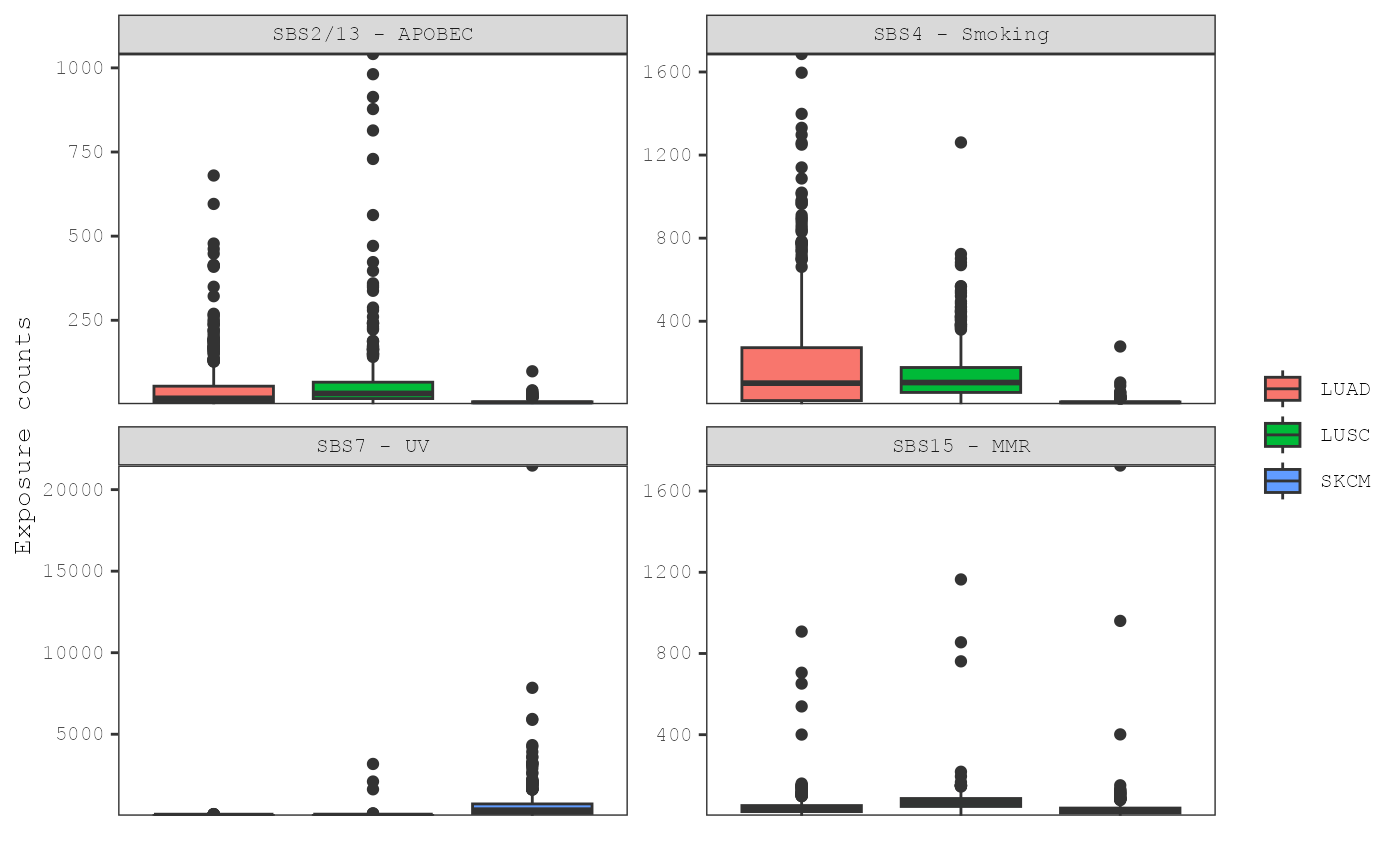
plot_exposures(musica_filter, "ex_result",
plot_type = "box", group_by = "signature",
color_by = "annotation", annotation = "Tumor_Type"
)
To verify that the deconvolution algorithm produced good signatures,
one strategy is to examine the patterns of mutations in individual
samples with a high predicted percentage of a particular signature. If
the shape of the counts match the patterns of the signature, then this
is a good indicator that the deconvolution algorithm worked well. Counts
for individual samples can be plotted with the
plot_sample_counts function. For example, we can plot the
sample with the highest proportion of the APOBEC signature:
# Normalize exposures
expos.prop <- prop.table(expos, margin = 2)
# Plot counts for the sample with the higest level of exposures for sigs #2
# and #4
ix <- c(which.max(expos.prop[2, ]), which.max(expos.prop[4, ]))
plot_sample_counts(musica_filter, sample_names = colnames(expos.prop)[ix],
modality = "SBS96")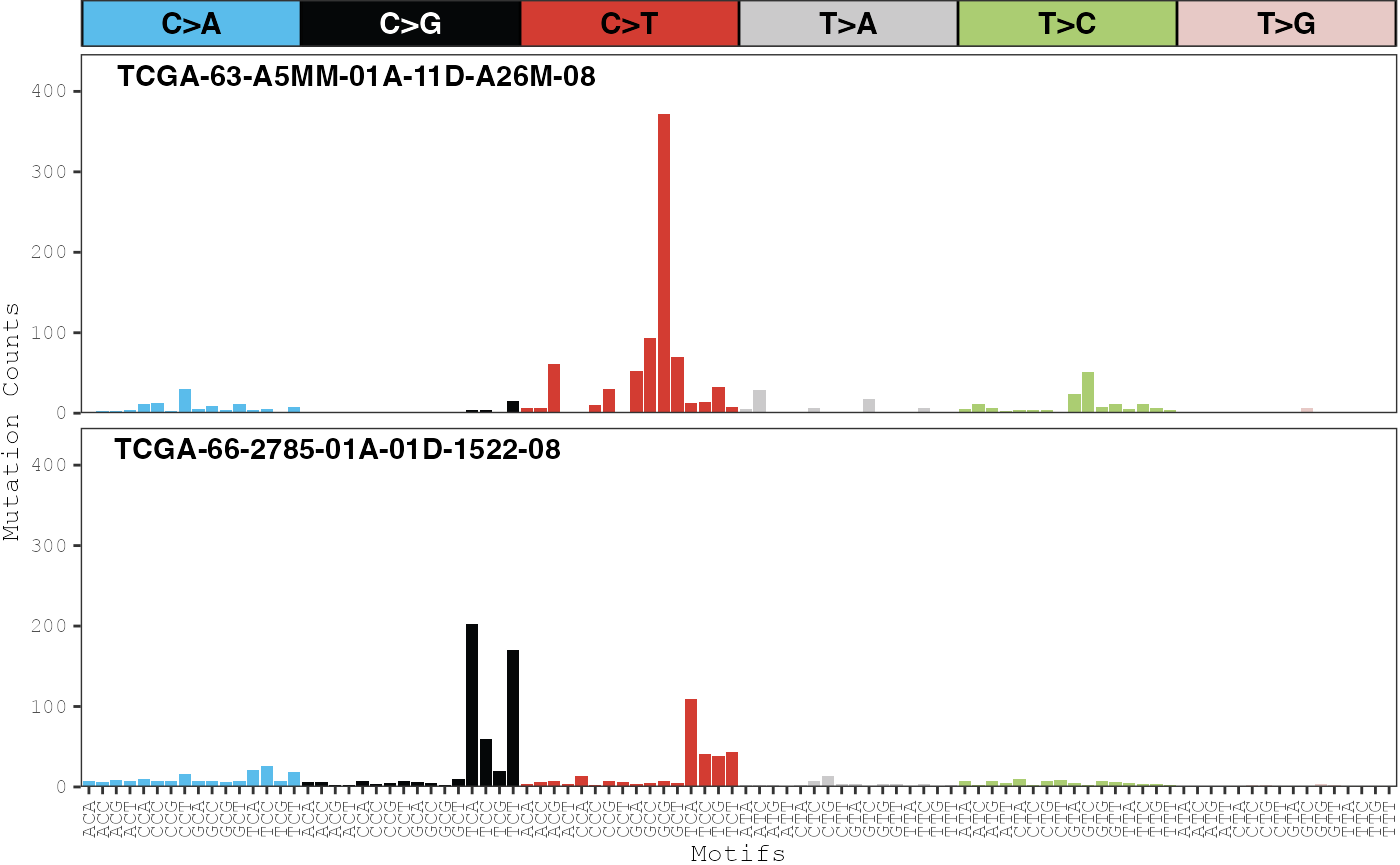
Predict exposures from existing signatures
Predict COSMIC signatures
Instead of discovering mutational signatures and exposures from a
dataset de novo, a better result may be obtained by predicting
the exposures of signatures that have been previously estimated in other
datasets. Predicting exposures for pre-existing signatures may have more
sensitivity for detecting active compared to the discovery-based methods
as we are incorporating prior information derived from larger datasets.
The musicatk package incorporates several methods for
estimating exposures given a set of pre-existing signatures. For
example, the exposures for COSMIC signatures 1, 4, 7, 13, and 15 can be
predicted in our current dataset. Note that we are including COSMIC
signature 1 in the prediction even though it did not show up in the
discovery algorithm as this signature has been previously shown to be
active in lung tumors and we are also including both APOBEC signatures
(2 and 13) which were previously combined into 1 signature in the
discovery method.
# Load COSMIC V2 data
data("cosmic_v2_sigs")
# Predict pre-existing exposures using the "lda" method
predict_exposure(
musica = musica_filter, modality = "SBS96",
signature_res = cosmic_v2_sigs,
model_id = "result_cosmic_selected_sigs",
signatures_to_use = c(1, 2, 4, 6, 7, 13), algorithm = "lda"
)
# Plot exposures
plot_exposures(musica_filter, "result_cosmic_selected_sigs", plot_type = "bar",
num_samples = 50)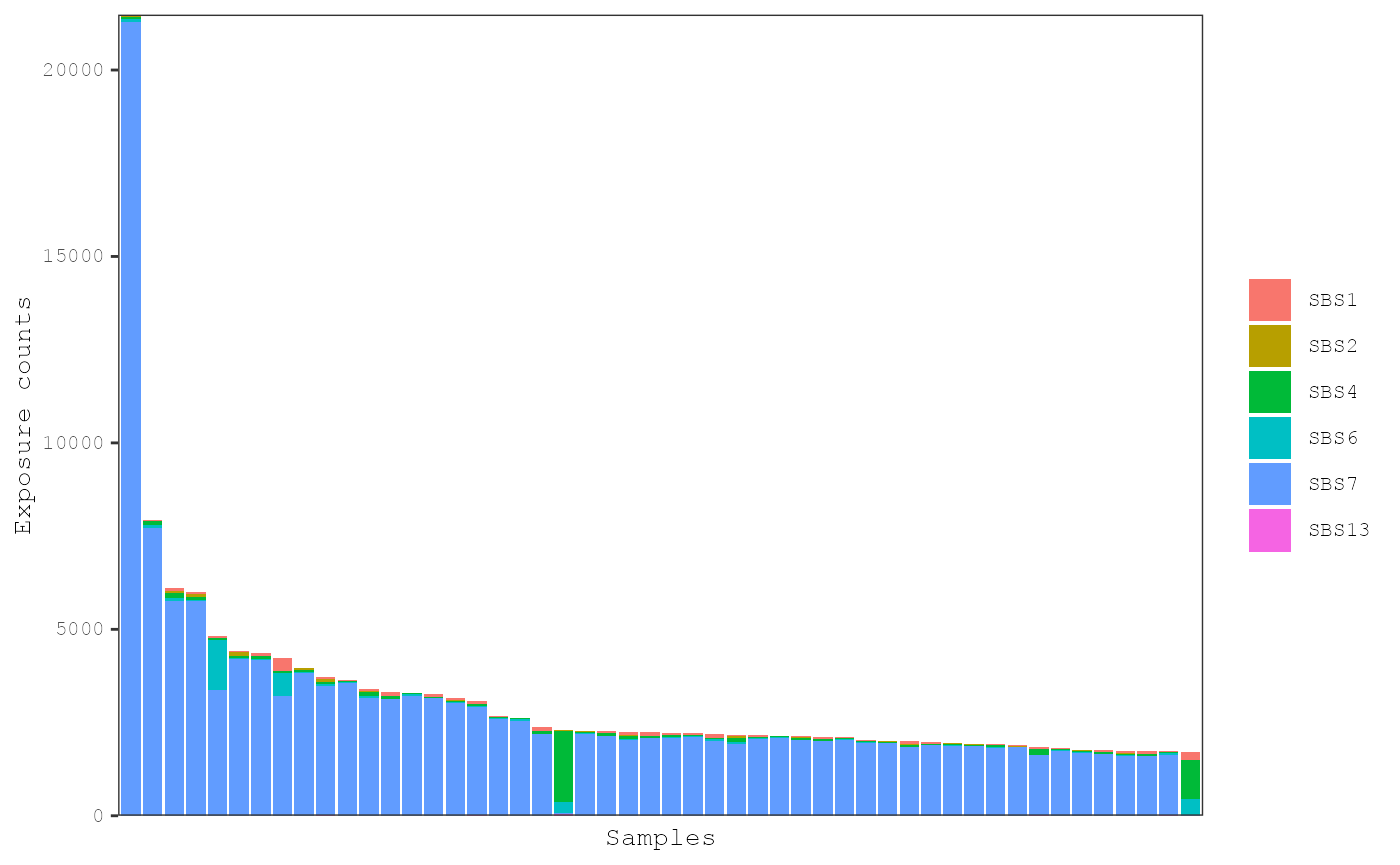
The cosmic_v2_sigs object is just a
result_model object containing COSMIC V2 signatures without
any sample or exposure information. Note that if
signatures_to_use is not supplied by the user, then
exposures for all signatures in the result object will be estimated. Any
result_model object can be given to the
signature_res parameter. Exposures can be predicted for
samples in any musica object from any
result_model object as long as the same mutation schema was
utilized.
Prediction with signature selection
In many cases, researchers will not know the signatures that are
active in a cohort of samples beforehand. While it would be easy to
predict all COSMIC signatures, this can have detrimental effects on the
output. Including signatures not actually active in the cohort of
samples may introduce additional noise in the estimates for the
exposures for the signatures that are truly present in the dataset.
Additionally, including extra signatures may induce a false signal for
the exposures of the non-active signatures. The musicatk
package has a “two-step” prediction process. In the first step,
exposures for all signatures will be estimated. Then a subset of
signatures will be selected as “active” in the dataset and only the
exposures for the active signatures will be estimated. This two-step
process can be done automatically using the
auto_predict_grid function:
# Predict exposures with auto selection of signatures
auto_predict_grid(musica_filter, modality = "SBS96",
signature_res = cosmic_v2_sigs, algorithm = "lda",
model_id = "result_cosmic_auto",
sample_annotation = "Tumor_Type")## LUAD## LUSC## SKCM
# See list of selected signatures
rownames(exposures(musica_filter, "result", "SBS96", "result_cosmic_auto"))## [1] "SBS1" "SBS13" "SBS15" "SBS16" "SBS18" "SBS2" "SBS21" "SBS24" "SBS26"
## [10] "SBS3" "SBS4" "SBS6" "SBS7"In this result, 13 of the 30 original COSMIC V2 signatures were
selected including several signatures that were not previously included
in our first prediction with manually selected signatures. If multiple
groups of samples are present in the dataset that are expected to have
somewhat different sets of active signatures (e.g. multiple tumor
types), then this 2-step process can be improved by performing signature
selection within each group. This can be achieved by supplying the
sample_annoation parameter. In our example, exposures were
predicted in the three different tumor types by supplying the
Tumor_Type annotation to sample_annotation.
This parameter can be left NULL if no grouping annotation
is available.
The three major parameters that determine whether a signature is present in a dataset on the first pass are:
-
min_exists- A signature will be considered active in a sample if its exposure level is above this threshold (Default0.05). -
proportion_samples- A signature will be considered active in a cohort and included in the second pass if it is active in at least this proportion of samples (Default0.25). -
rare_exposure- A signature will be considered active in a cohort and included in the second pass if the proportion of its exposure is above this threshold in at least one sample (Default0.4). This parameter is meant to capture signatures that produce high number of mutations but are found in a small number of samples (e.g. Mismatch repair).
Assess predicted signatures
It is almost always worthwhile to manually assess and confirm the signatures predicted to be present within a dataset, especially for signatures that have similar profiles to one another. For example, both COSMIC Signature 4 (smoking) and Signature 24 (aflatoxin) were predicted to be present within our dataset. The smoking-related signature is expected as our cohort contains lung cancers, but the aflatoxin signature is unexpected given that it is usually found in liver cancers. These signatures both have a strong concentration of C>A tranversions. In fact, we can see that the predicted exposures for these signatures are highly correlated to each other across samples:
{kind=link}
{kind=link}
e <- exposures(musica_filter, "result", "SBS96", "result_cosmic_auto")
plot(e["SBS4", ], e["SBS24", ], xlab = "SBS4", ylab = "SBS24")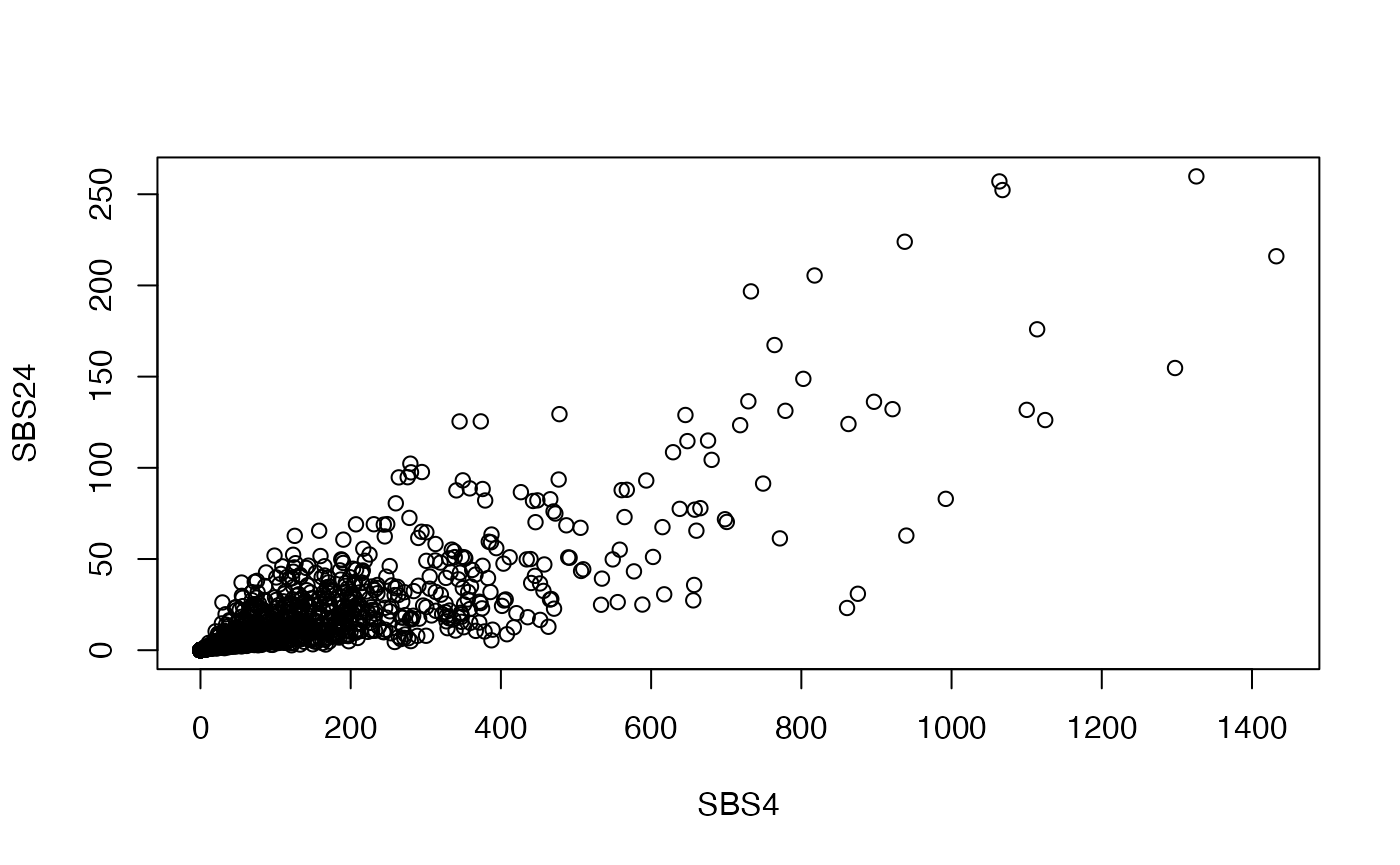
Therefore, we will want to remove Signature 24 from our final prediction model. Signature 18 is another one with a high prevalence of C>A transversion at specific trinucleotide contexts. However, at least a few samples have high levels of Signature 18 without correspondingly high levels of Signature 4:
{kind=link}
plot(e["SBS4", ], e["SBS18", ], xlab = "SBS4", ylab = "SBS18")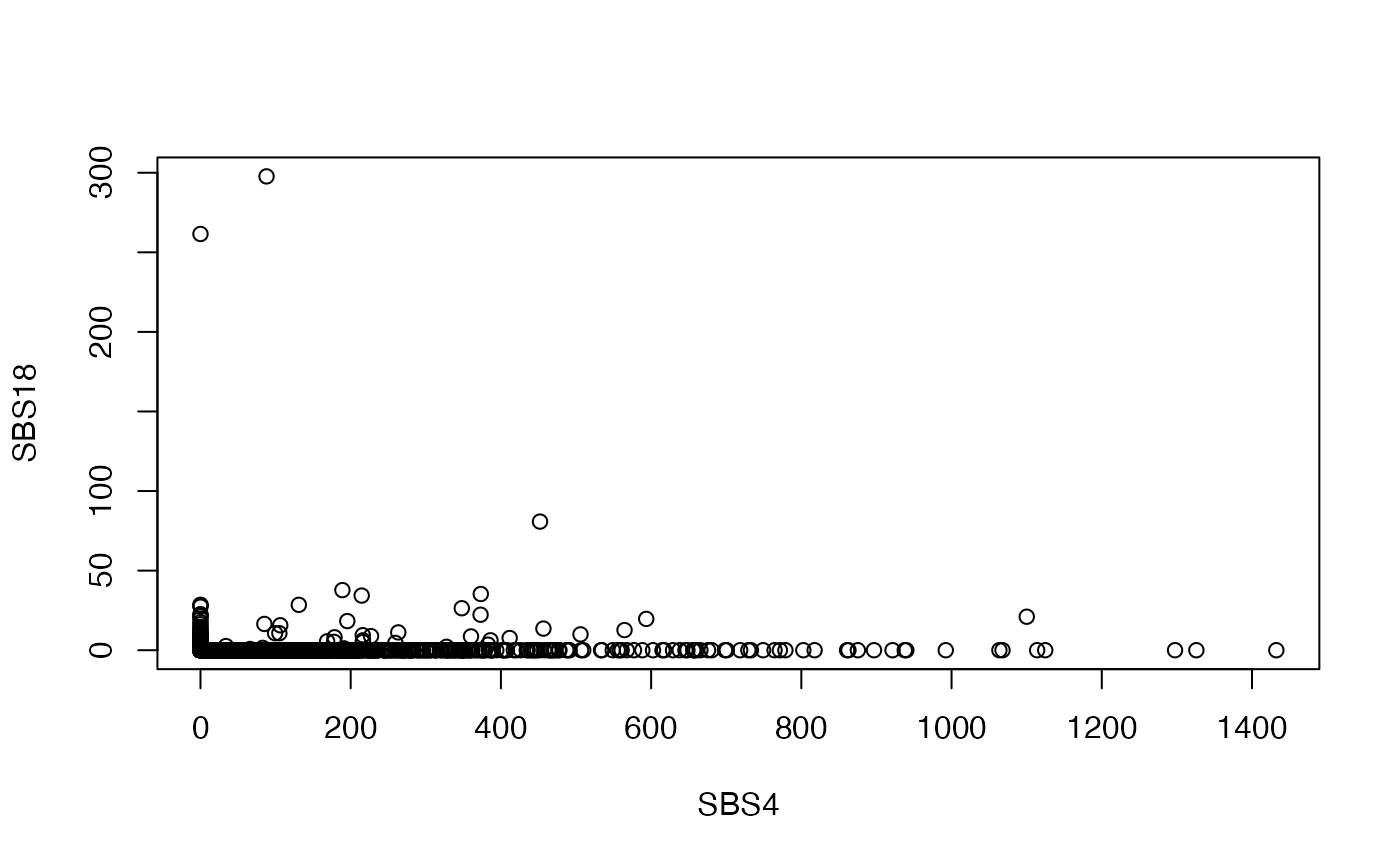
plot_exposures(musica_filter, "result_cosmic_auto", num_samples = 25,
sort_samples = "SBS18")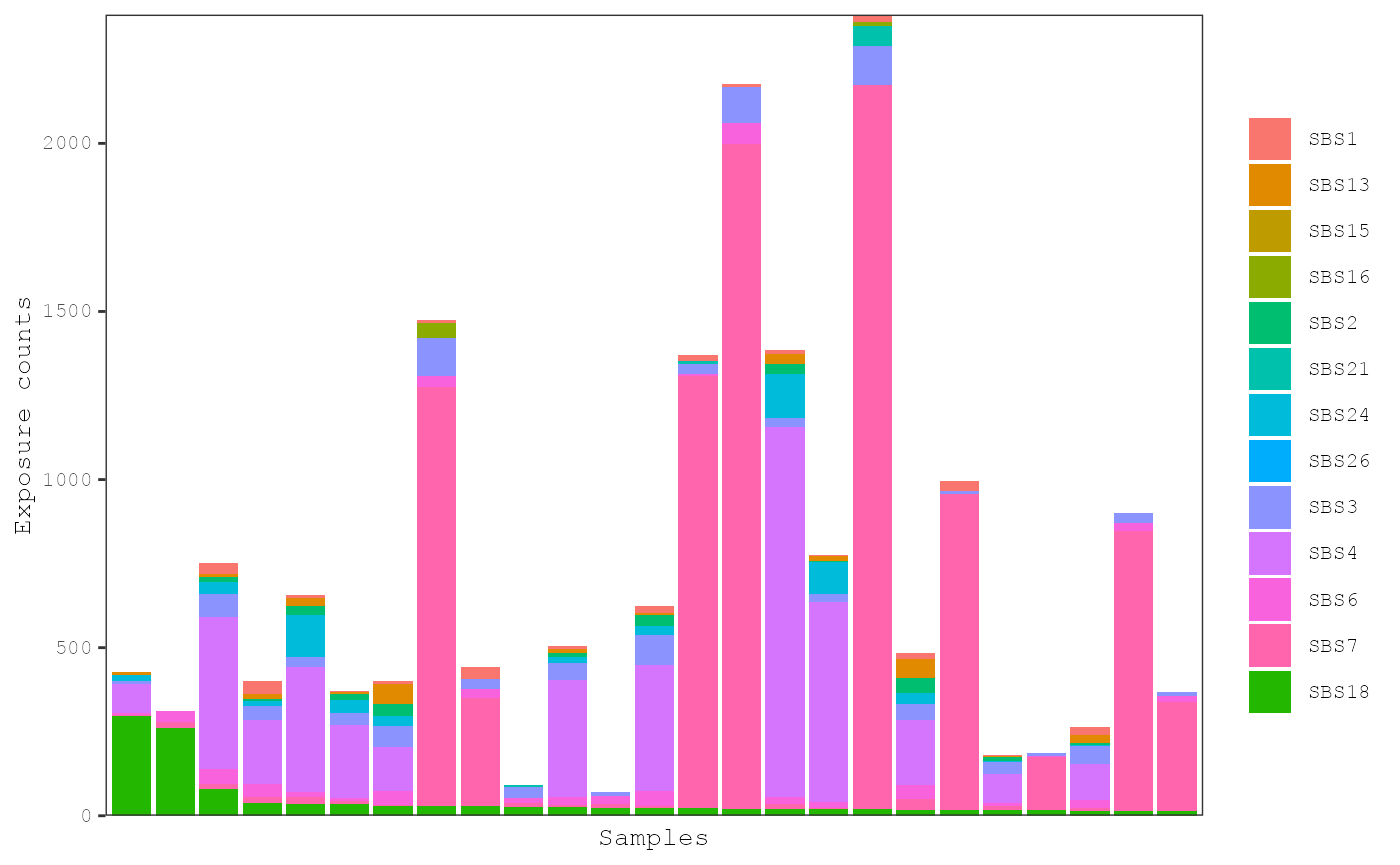
Additionally, 2 of the 3 samples are skin cancers where the smoking signature is not usually expected:
## Tumor_Type ID
## TCGA-37-5819-01A-01D-1632-08 "LUSC" "TCGA-37-5819-01A-01D-1632-08"
## TCGA-EE-A184-06A-11D-A196-08 "SKCM" "TCGA-EE-A184-06A-11D-A196-08"
## TCGA-22-5472-01A-01D-1632-08 "LUSC" "TCGA-22-5472-01A-01D-1632-08"As a final check, we can look at the counts of the individual samples with high levels of Signature 18:
plot_sample_counts(musica_filter, sample_names = "TCGA-ER-A19P-06A-11D-A196-08")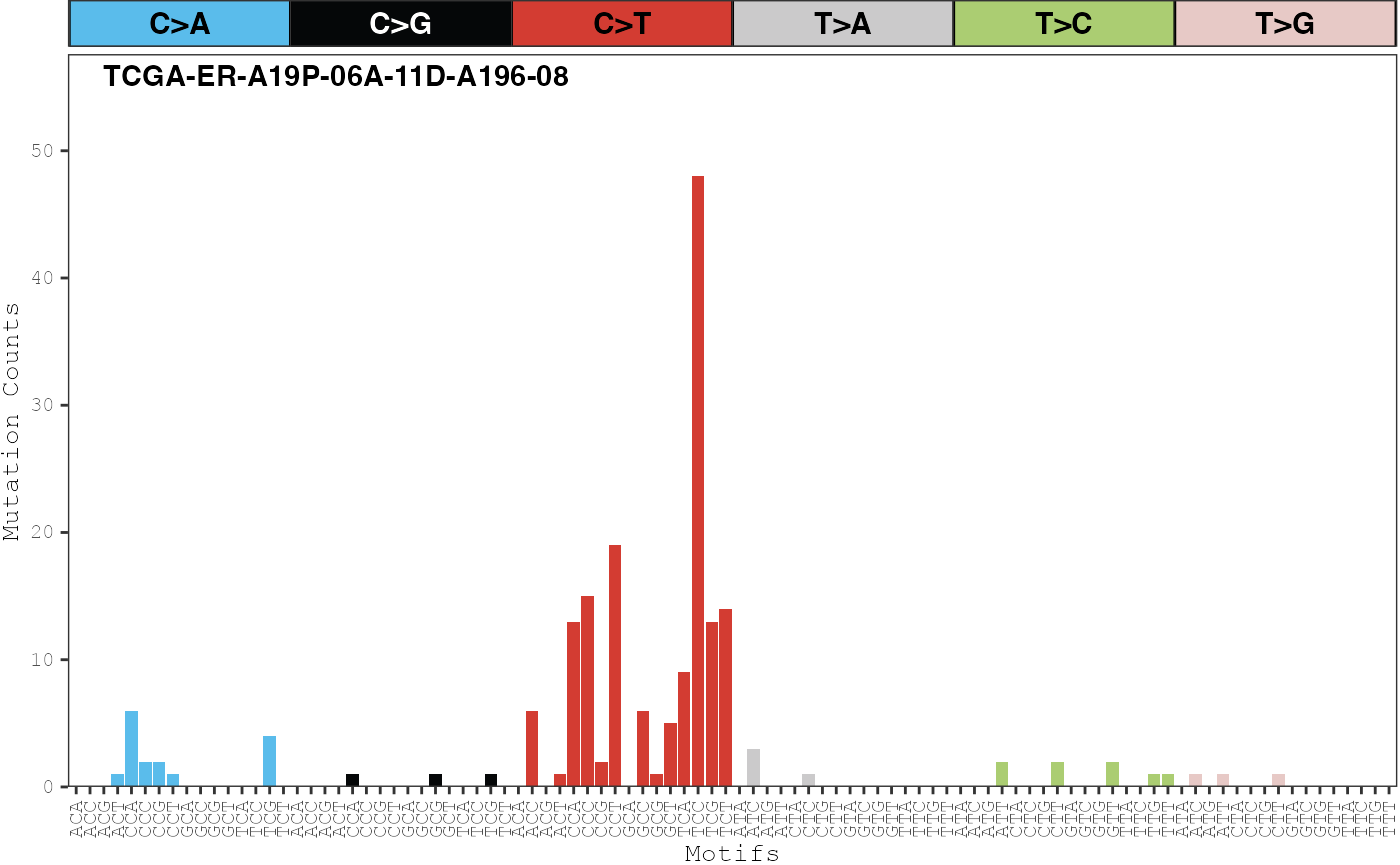
This sample clearly has high levels of both the UV signature confirming that it is likely a skin cancer. Signature 18 is also likely to be active as a high number of C>A mutations at CCA, TCA, and TCT trinucleotide contexts can be observed. Given these results, Signature 18 will be kept in the final analysis.
After additional analysis of other signatures, we also want to remove
Signature 3 as that is predominantly found in tumors with BRCA
deficiencies (e.g. breast cancer) and in samples with high rates of
indels (which are not observed here). The predict_exposure
function will be run one last time with the curated list of signatures
and this final result will be used in the rest of the down-stream
analyses:
# Predict pre-existing exposures with the revised set of selected signatures
predict_exposure(musica = musica_filter, modality = "SBS96",
signature_res = cosmic_v2_sigs,
signatures_to_use = c(1, 2, 4, 6, 7, 13, 15, 18, 26),
model_id = "result_cosmic_final", algorithm = "lda")Downstream analyses
Visualize relationships between samples with 2-D embedding
The create_umap function embeds samples in 2 dimensions
using the umap function from the uwot package.
The major parameters for fine tuning the UMAP are
n_neighbors, min_dist, and
spread. Generally, a higher min_dist will
create more separation between the larger groups of samples while a
lower See ?uwot::umap for more information on these
parameters as well as this tutorial for
fine-tuning. Here, a UMAP will be created with standard parameters:
set.seed(1)
create_umap(musica_filter, "result_cosmic_final")Note that while we are using the result_cosmic_final
model which came from the prediction algorithm, we could have also used
the ex_result model generated by the discovery algorithm.
The plot_umap function will generate a scatter plot of the
UMAP coordinates. The points of plot will be colored by the level of a
signature by default:
plot_umap(musica_filter, "result_cosmic_final")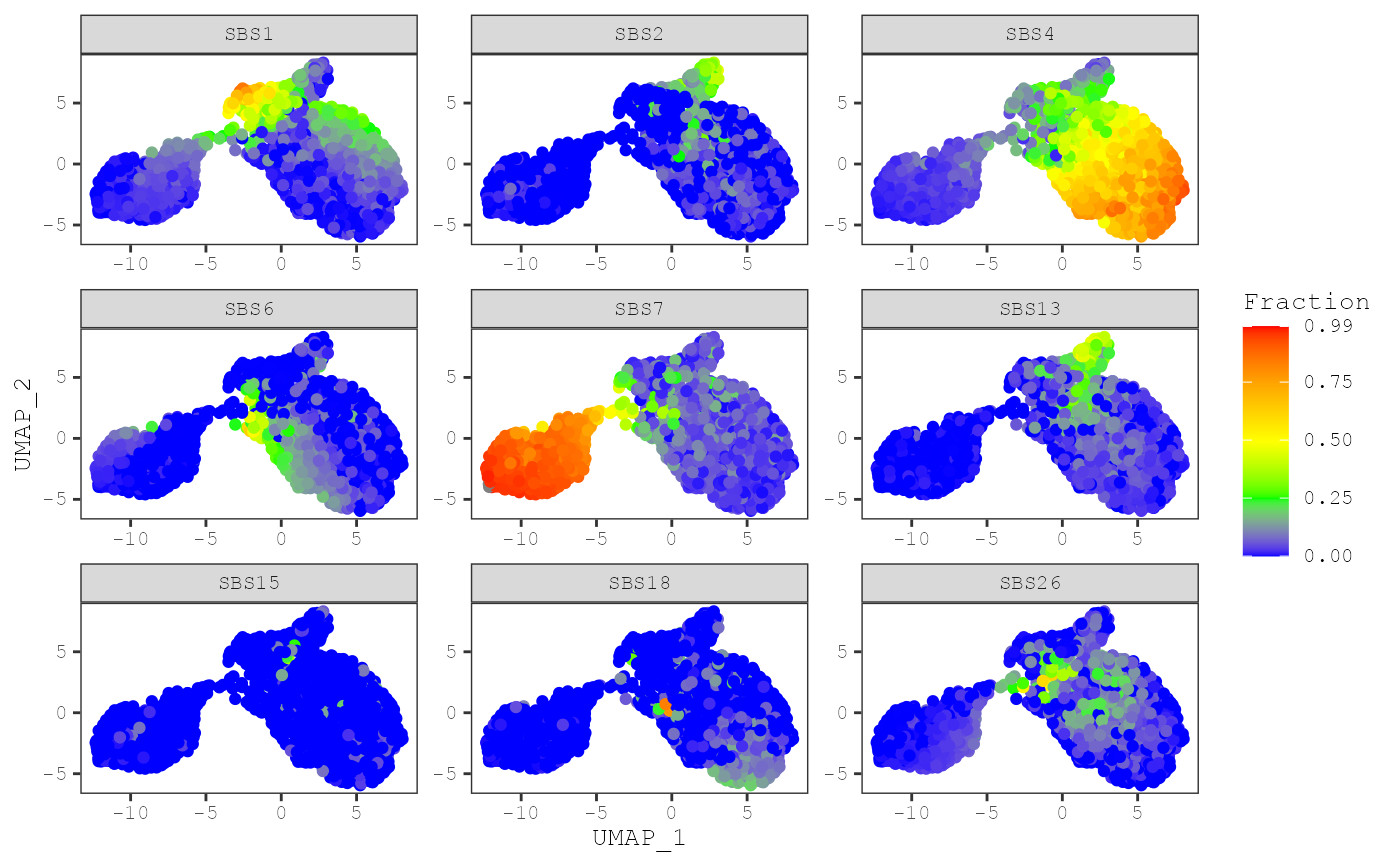
By default, the exposures for each sample will share the same color
scale. However, exposures for some signatures may have really high
levels compared to others. To make a plot where exposures for each
signature will have their own color scale, you can set
same_scale = FALSE:
plot_umap(musica_filter, "result_cosmic_final", same_scale = FALSE)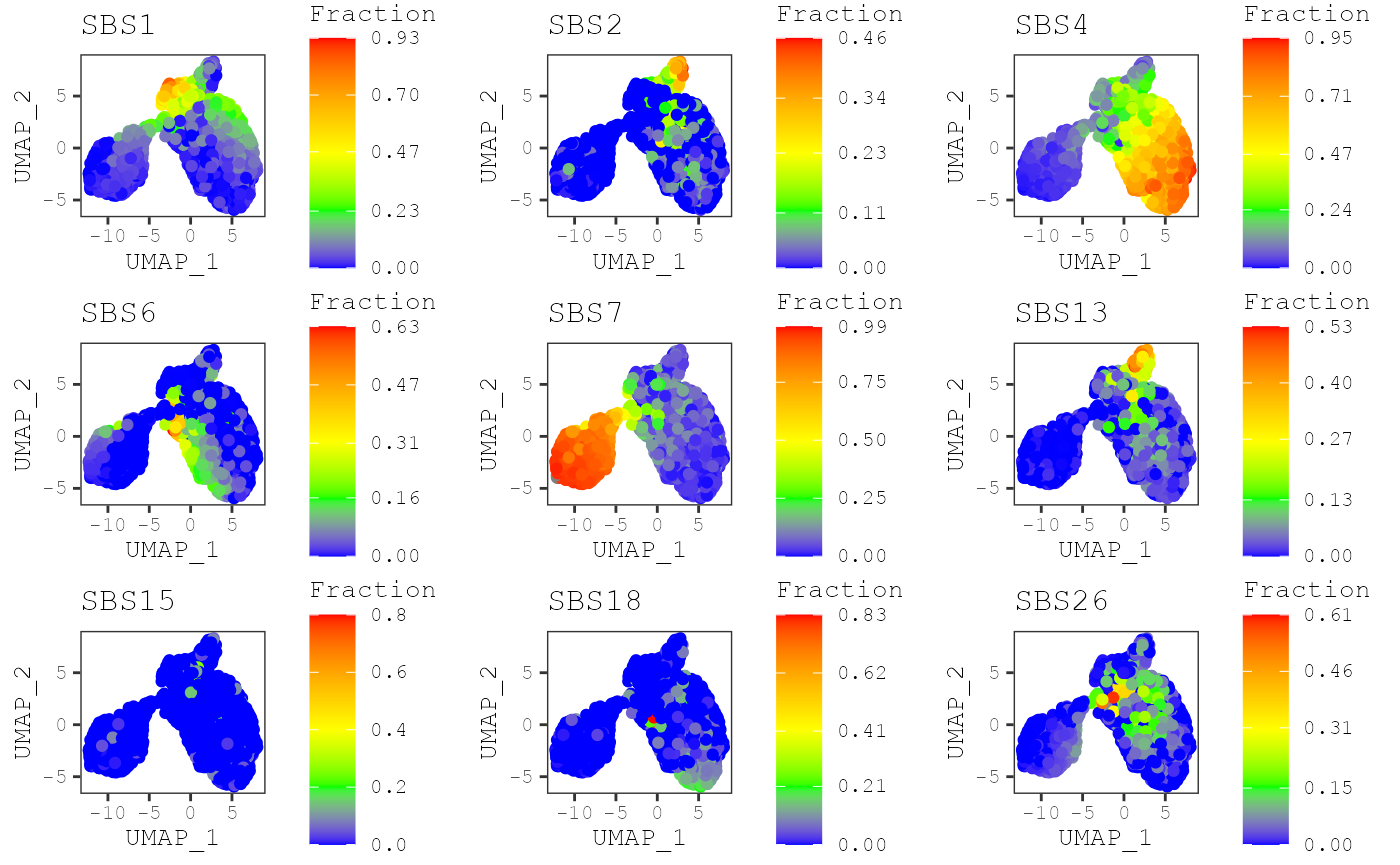
Lastly, points can be colored by a Sample Annotation by setting
color_by = "annotation" and the annotation
parameter to the name of the annotation:
plot_umap(musica_filter, "result_cosmic_final", color_by = "annotation",
annotation = "Tumor_Type")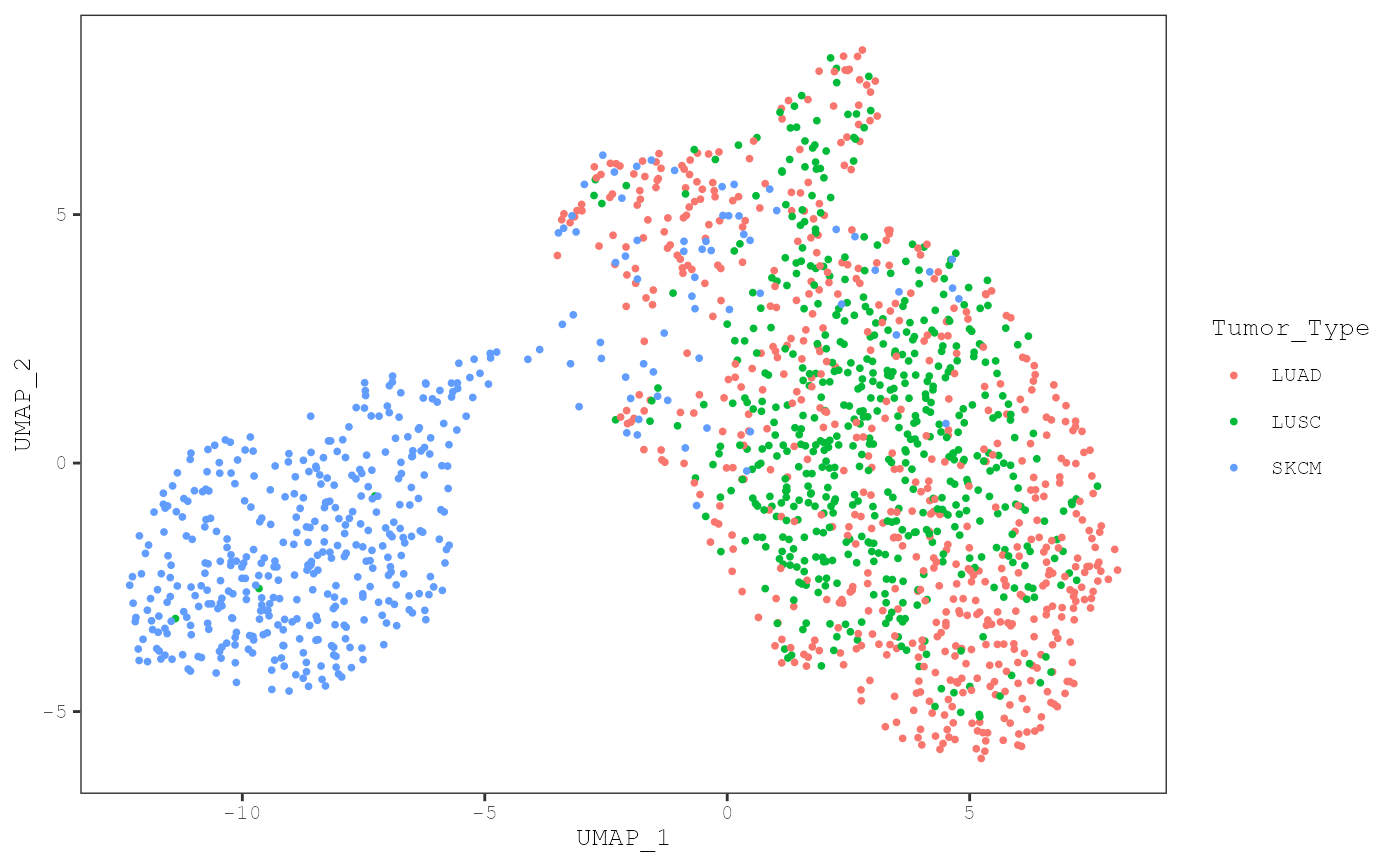
If we set add_annotation_labels = TRUE, the centroid of
each group is identified using medians and the labels are plotted at the
position of the centroid:
plot_umap(musica_filter, "result_cosmic_final", color_by = "annotation",
annotation = "Tumor_Type", add_annotation_labels = TRUE)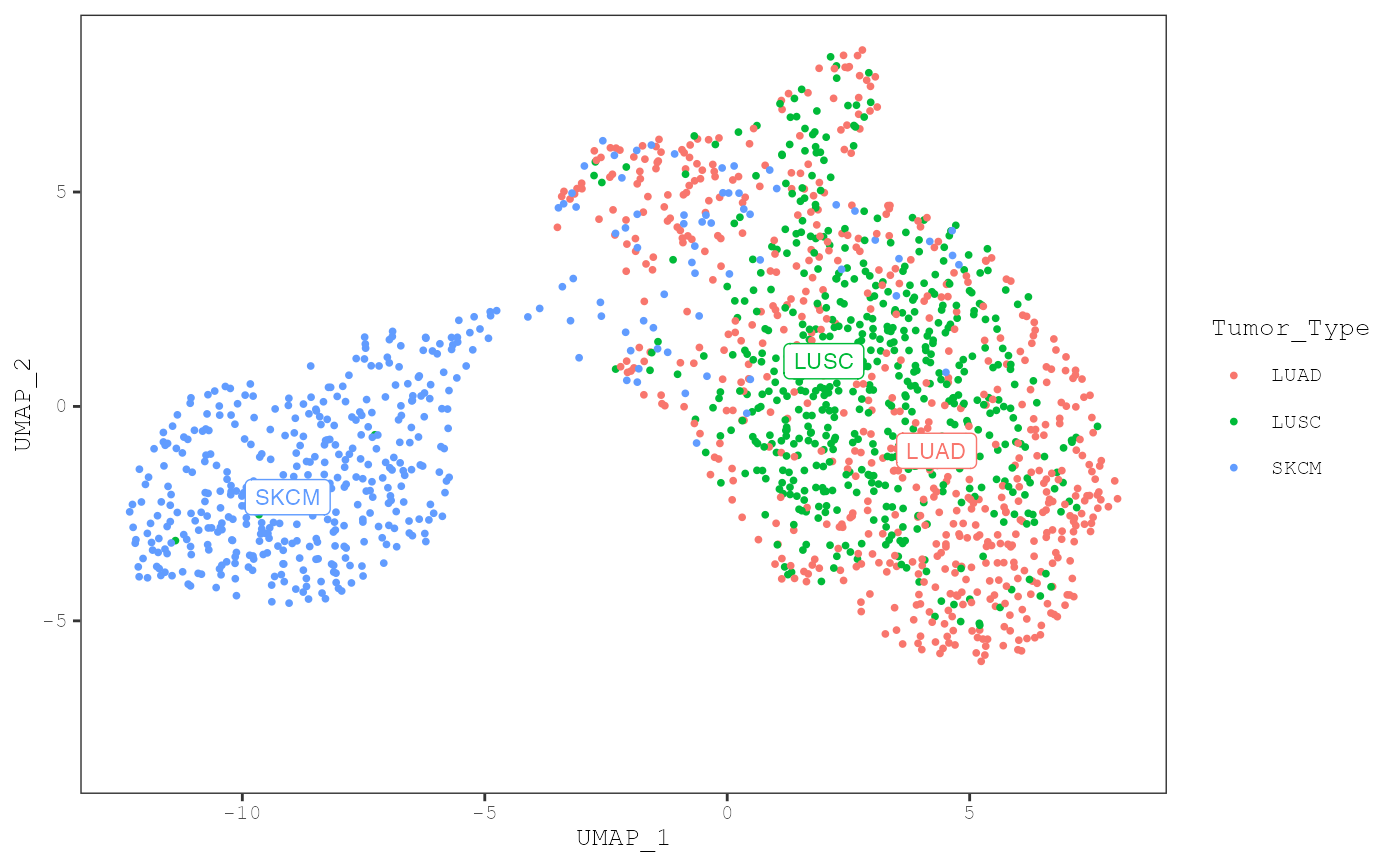
Plotting exposures in a heatmap
Exposures can be displayed in a heatmap where each row corresponds to a siganture and each column correponds to a sample:
plot_heatmap(musica_filter, "result_cosmic_final")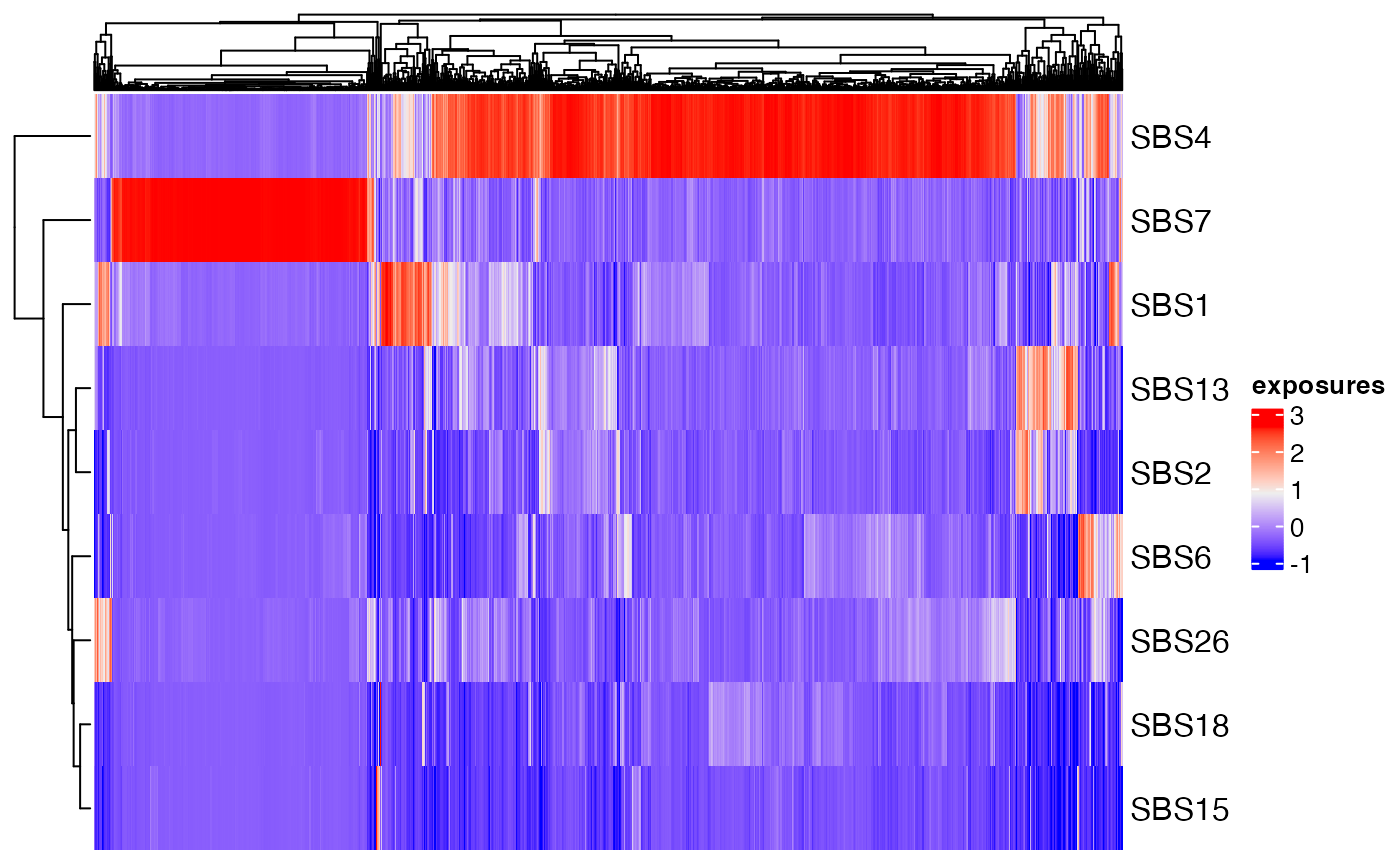
By default, signatures are scaled to have a mean of zero and a
standard deviation of 1 across samples (i.e. z-scored). This can be
turned off by setting scale = FALSE. Sample annotations can
be displayed in the column color bar by setting the
annotation parameter:
plot_heatmap(musica_filter, "result_cosmic_final", annotation = "Tumor_Type")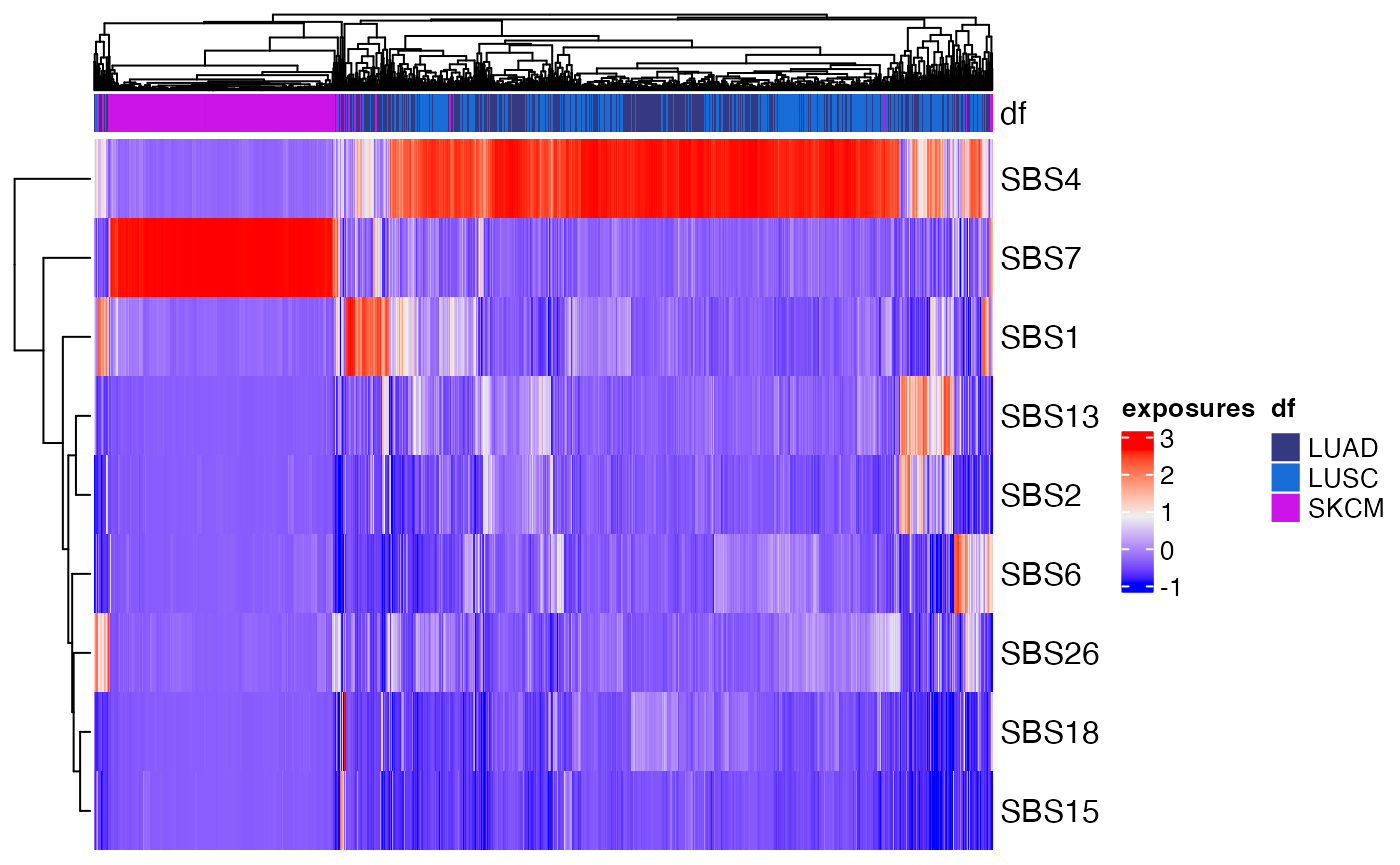
The heatmap shows that Signature 4 and Signature 7 are largely mutually exclusive from one another and can be used to separate lung and skin cancers. Additionally, subsets of signatures or samples can be displayed. For example, if we only want to examine signatures involved in mismatch repair, we can select signatures 6, 15, and 26:
plot_heatmap(musica_filter, "result_cosmic_final", annotation = "Tumor_Type",
subset_signatures = c("SBS6", "SBS15", "SBS26"))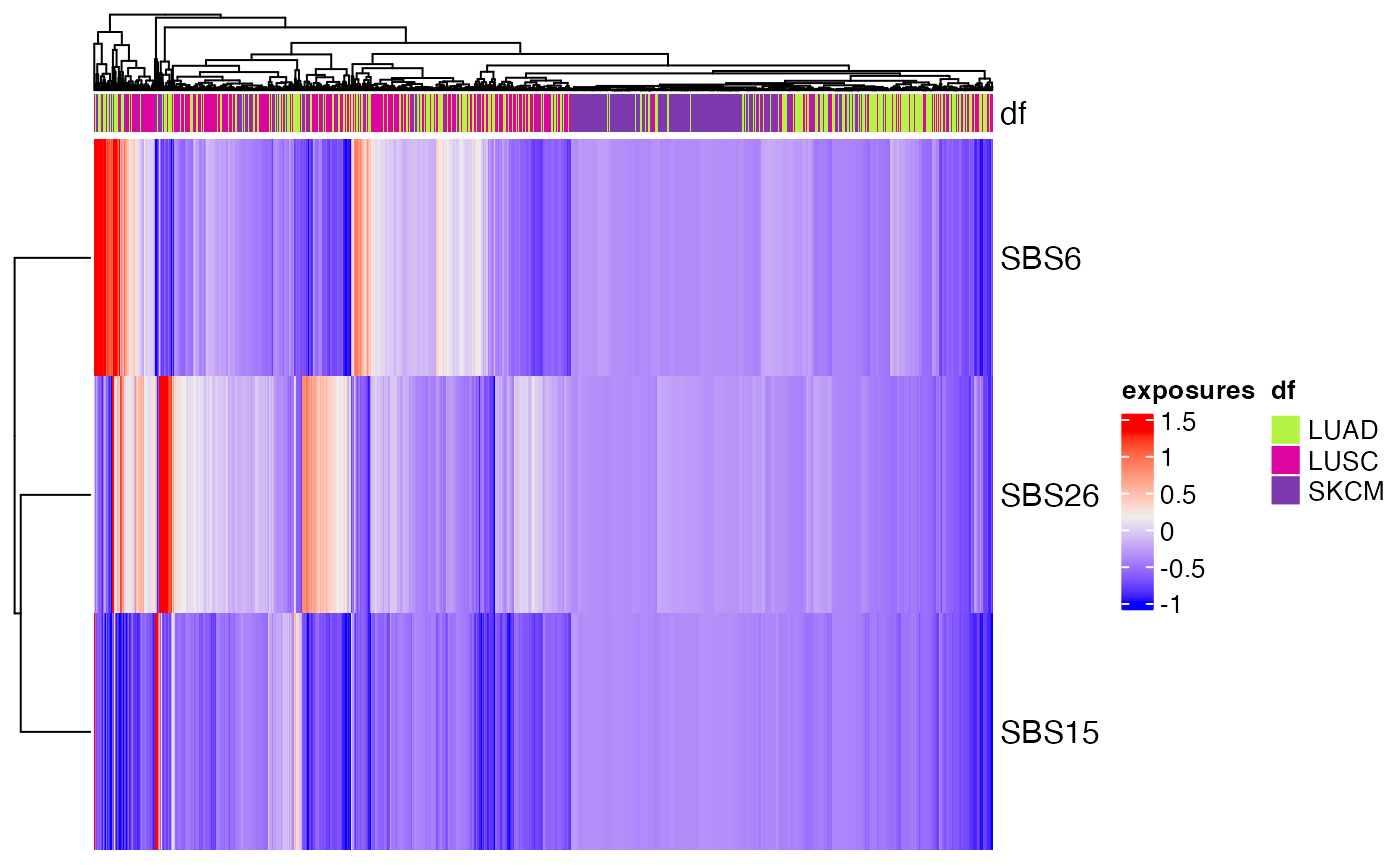
In this heatmap, we can see that only a small subset of distinct samples have relatively higher levels of these signatures.
Clustering samples based on exposures
Samples can be grouped into de novo clusters using a
several algorithms from the factoextra and cluster packages such as
pam or kmeans. One major challenge is choosing
the number of clusters (k). The function k_select has
several metrics for examining cluster stability such as total within
cluster sum of squares (wss), Silhouette Width
(silhouette), and the Gap Statistic
(gap_stat).
k_select(musica_filter, "result_cosmic_final", method = "silhouette",
clust.method = "pam", n = 20)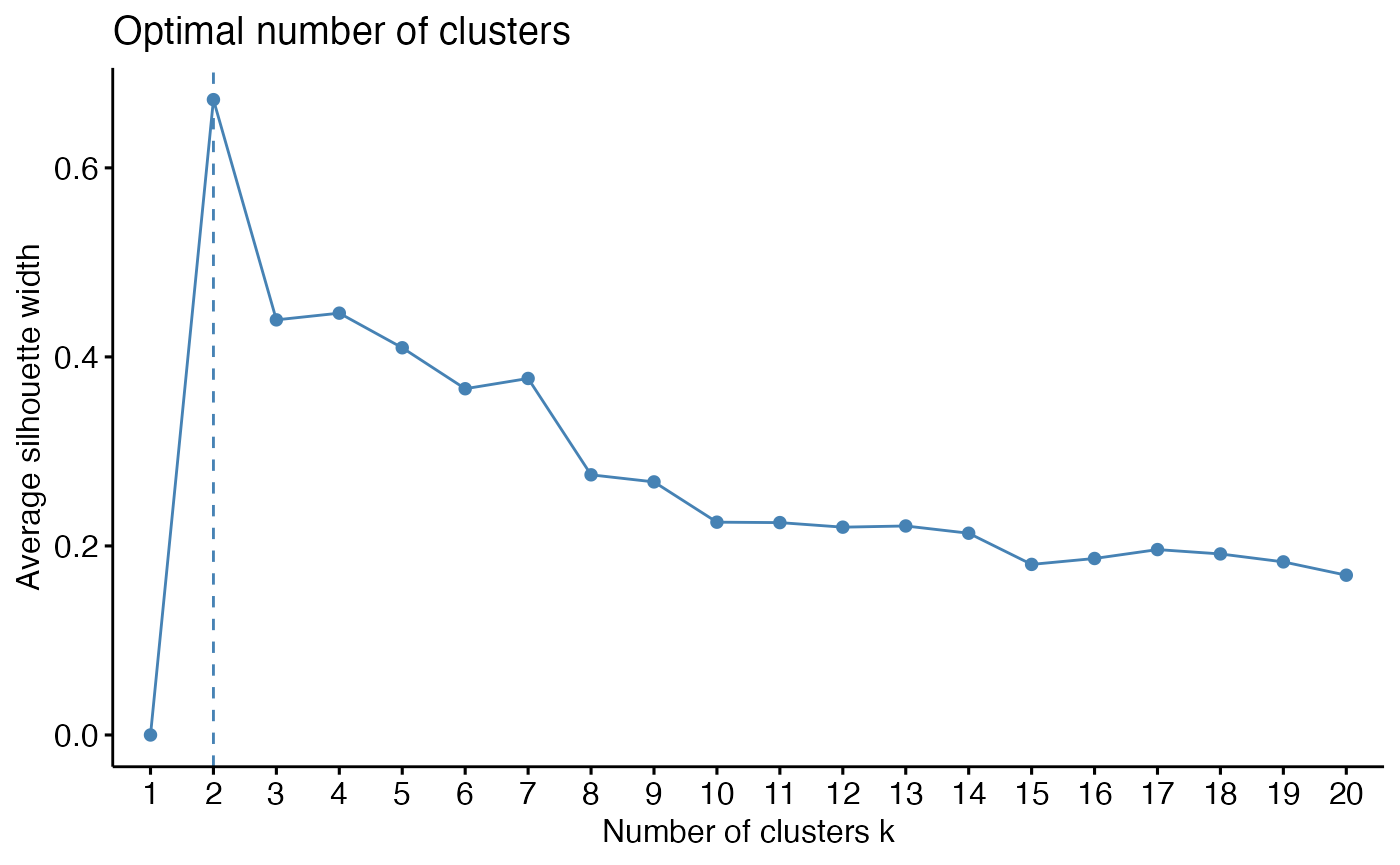
While 2 clusters may be the most optimal choice, this would just
correspond to the two large clusters of lung and skin tumors. Therefore,
choosing a higher value may be more informative. The next major drop in
the silhouette width is after k = 6, so we will select this
moving forward and perform the clustering:
clusters <- cluster_exposure(musica_filter, "result_cosmic_final",
method = "pam", nclust = 6)## Metric: 'euclidean'; comparing: 1614 vectors.Clusters can be visualized on the UMAP with the
plot_cluster function:
clusters[, 1] <- as.factor(clusters[, 1])
plot_cluster(musica_filter, "result_cosmic_final", cluster = clusters,
group = "none")Additional features
Plotly for interactive plots
The functions plot_signatures,
plot_exposures, and plot_umap have the ability
to create ggplotly
plots by simply specifying plotly = TRUE. Plotly plots are
interactive and allow users to zoom and re-sizing plots, turn on and off
annotation types and legend values, and hover over elements of the plots
(e.g. bars or points) to more information about that element
(e.g. sample name). Here are examples of plot_signatures
and plot_exposures
plot_signatures(musica_filter, "result_cosmic_final", plotly = TRUE)
plot_exposures(musica_filter, "result_cosmic_final", num_samples = 25,
plotly = TRUE)COSMIC signatures annotated to be active in a tumor type
The signatures predicted to be present in each tumor type according to the COSMIC V2 database can be quickly retrieved. For example, we can find which signatures are predicted to be present in lung cancers:
cosmic_v2_subtype_map("lung")## lung adeno## 124561317## lung small cell## 14515## lung squamous## 124513Creating custom tables
Custom count tables can be created from user-defined mutation-level
annotations using the build_custom_table function.
# Adds strand information to the 'variant' table
annotate_transcript_strand(musica, genome_build = "hg38", build_table = FALSE)## 2135 genes were dropped because they have exons located on both strands
## of the same reference sequence or on more than one reference sequence,
## so cannot be represented by a single genomic range.
## Use 'single.strand.genes.only=FALSE' to get all the genes in a
## GRangesList object, or use suppressMessages() to suppress this message.
# Generates a count table from strand
build_custom_table(
musica = musica, variant_annotation = "Transcript_Strand",
name = "Transcript_Strand",
description = "A table of transcript strand of variants",
data_factor = c("T", "U"), overwrite = TRUE
)Session Information
## R version 4.4.0 (2024-04-24)
## Platform: aarch64-apple-darwin20
## Running under: macOS Ventura 13.1
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## time zone: America/New_York
## tzcode source: internal
##
## attached base packages:
## [1] stats graphics grDevices datasets utils methods base
##
## other attached packages:
## [1] TCGAbiolinks_2.32.0 musicatk_2.0.0 NMF_0.27
## [4] Biobase_2.64.0 BiocGenerics_0.50.0 cluster_2.1.6
## [7] rngtools_1.5.2 registry_0.5-1
##
## loaded via a namespace (and not attached):
## [1] RcppAnnoy_0.0.22
## [2] BiocIO_1.14.0
## [3] bitops_1.0-7
## [4] filelock_1.0.3
## [5] matrixTests_0.2.3
## [6] tibble_3.2.1
## [7] R.oo_1.26.0
## [8] XML_3.99-0.17
## [9] factoextra_1.0.7
## [10] lifecycle_1.0.4
## [11] httr2_1.0.2
## [12] rstatix_0.7.2
## [13] doParallel_1.0.17
## [14] NLP_0.2-1
## [15] lattice_0.22-6
## [16] vroom_1.6.5
## [17] MASS_7.3-61
## [18] crosstalk_1.2.1
## [19] backports_1.5.0
## [20] magrittr_2.0.3
## [21] plotly_4.10.4
## [22] sass_0.4.9
## [23] rmarkdown_2.27
## [24] jquerylib_0.1.4
## [25] yaml_2.3.9
## [26] BSgenome.Hsapiens.UCSC.hg38_1.4.5
## [27] topicmodels_0.2-16
## [28] cowplot_1.1.3
## [29] DBI_1.2.3
## [30] RColorBrewer_1.1-3
## [31] abind_1.4-5
## [32] zlibbioc_1.50.0
## [33] rvest_1.0.4
## [34] GenomicRanges_1.56.1
## [35] purrr_1.0.2
## [36] R.utils_2.12.3
## [37] RCurl_1.98-1.16
## [38] VariantAnnotation_1.50.0
## [39] rappdirs_0.3.3
## [40] circlize_0.4.16
## [41] GenomeInfoDbData_1.2.12
## [42] IRanges_2.38.1
## [43] S4Vectors_0.42.1
## [44] ggrepel_0.9.5
## [45] tm_0.7-13
## [46] irlba_2.3.5.1
## [47] BiocStyle_2.32.1
## [48] conclust_1.1
## [49] pkgdown_2.1.0
## [50] codetools_0.2-20
## [51] DelayedArray_0.30.1
## [52] xml2_1.3.6
## [53] tidyselect_1.2.1
## [54] shape_1.4.6.1
## [55] UCSC.utils_1.0.0
## [56] farver_2.1.2
## [57] TCGAbiolinksGUI.data_1.24.0
## [58] matrixStats_1.3.0
## [59] stats4_4.4.0
## [60] BiocFileCache_2.12.0
## [61] GenomicAlignments_1.40.0
## [62] jsonlite_1.8.8
## [63] GetoptLong_1.0.5
## [64] iterators_1.0.14
## [65] systemfonts_1.1.0
## [66] foreach_1.5.2
## [67] tools_4.4.0
## [68] progress_1.2.3
## [69] TxDb.Hsapiens.UCSC.hg19.knownGene_3.2.2
## [70] ragg_1.3.2
## [71] Rcpp_1.0.12
## [72] glue_1.7.0
## [73] gridExtra_2.3
## [74] SparseArray_1.4.8
## [75] xfun_0.45
## [76] MatrixGenerics_1.16.0
## [77] GenomeInfoDb_1.40.1
## [78] dplyr_1.1.4
## [79] withr_3.0.1
## [80] combinat_0.0-8
## [81] BiocManager_1.30.23
## [82] fastmap_1.2.0
## [83] MCMCprecision_0.4.0
## [84] fansi_1.0.6
## [85] digest_0.6.36
## [86] R6_2.5.1
## [87] textshaping_0.4.0
## [88] colorspace_2.1-0
## [89] gtools_3.9.5
## [90] biomaRt_2.60.1
## [91] RSQLite_2.3.7
## [92] R.methodsS3_1.8.2
## [93] utf8_1.2.4
## [94] tidyr_1.3.1
## [95] generics_0.1.3
## [96] renv_1.0.7
## [97] data.table_1.15.4
## [98] rtracklayer_1.64.0
## [99] prettyunits_1.2.0
## [100] httr_1.4.7
## [101] htmlwidgets_1.6.4
## [102] S4Arrays_1.4.1
## [103] uwot_0.2.2
## [104] pkgconfig_2.0.3
## [105] gtable_0.3.5
## [106] modeltools_0.2-23
## [107] blob_1.2.4
## [108] ComplexHeatmap_2.20.0
## [109] XVector_0.44.0
## [110] htmltools_0.5.8.1
## [111] carData_3.0-5
## [112] clue_0.3-65
## [113] scales_1.3.0
## [114] tidyverse_2.0.0
## [115] TxDb.Hsapiens.UCSC.hg38.knownGene_3.18.0
## [116] png_0.1-8
## [117] knitr_1.48
## [118] rstudioapi_0.16.0
## [119] tzdb_0.4.0
## [120] reshape2_1.4.4
## [121] rjson_0.2.21
## [122] curl_5.2.1
## [123] cachem_1.1.0
## [124] GlobalOptions_0.1.2
## [125] stringr_1.5.1
## [126] parallel_4.4.0
## [127] AnnotationDbi_1.66.0
## [128] restfulr_0.0.15
## [129] desc_1.4.3
## [130] pillar_1.9.0
## [131] grid_4.4.0
## [132] vctrs_0.6.5
## [133] slam_0.1-50
## [134] ggpubr_0.6.0
## [135] car_3.1-2
## [136] dbplyr_2.5.0
## [137] evaluate_0.24.0
## [138] readr_2.1.5
## [139] GenomicFeatures_1.56.0
## [140] cli_3.6.3
## [141] compiler_4.4.0
## [142] Rsamtools_2.20.0
## [143] rlang_1.1.4
## [144] crayon_1.5.3
## [145] ggsignif_0.6.4
## [146] labeling_0.4.3
## [147] plyr_1.8.9
## [148] fs_1.6.4
## [149] stringi_1.8.4
## [150] viridisLite_0.4.2
## [151] philentropy_0.8.0
## [152] gridBase_0.4-7
## [153] BiocParallel_1.38.0
## [154] munsell_0.5.1
## [155] Biostrings_2.72.1
## [156] lazyeval_0.2.2
## [157] Matrix_1.7-0
## [158] BSgenome_1.72.0
## [159] hms_1.1.3
## [160] bit64_4.0.5
## [161] ggplot2_3.5.1
## [162] KEGGREST_1.44.1
## [163] SummarizedExperiment_1.34.0
## [164] highr_0.11
## [165] broom_1.0.6
## [166] memoise_2.0.1
## [167] bslib_0.7.0
## [168] bit_4.0.5
## [169] downloader_0.4